Tumor mutation analysis¶
The Tumor mutation mnalysis (TMA) report builder filters, annotates, and summarizes variant data from tumor samples or from paired tumor versus normal sample pairs. TMA then adds annotation and applies user-defined filters to each variant. Variant annotation includes Variant Effect Prediction (VEP) impact and score, COSMIC annotation, relevant FDA approved drugs, and other publicly available data.
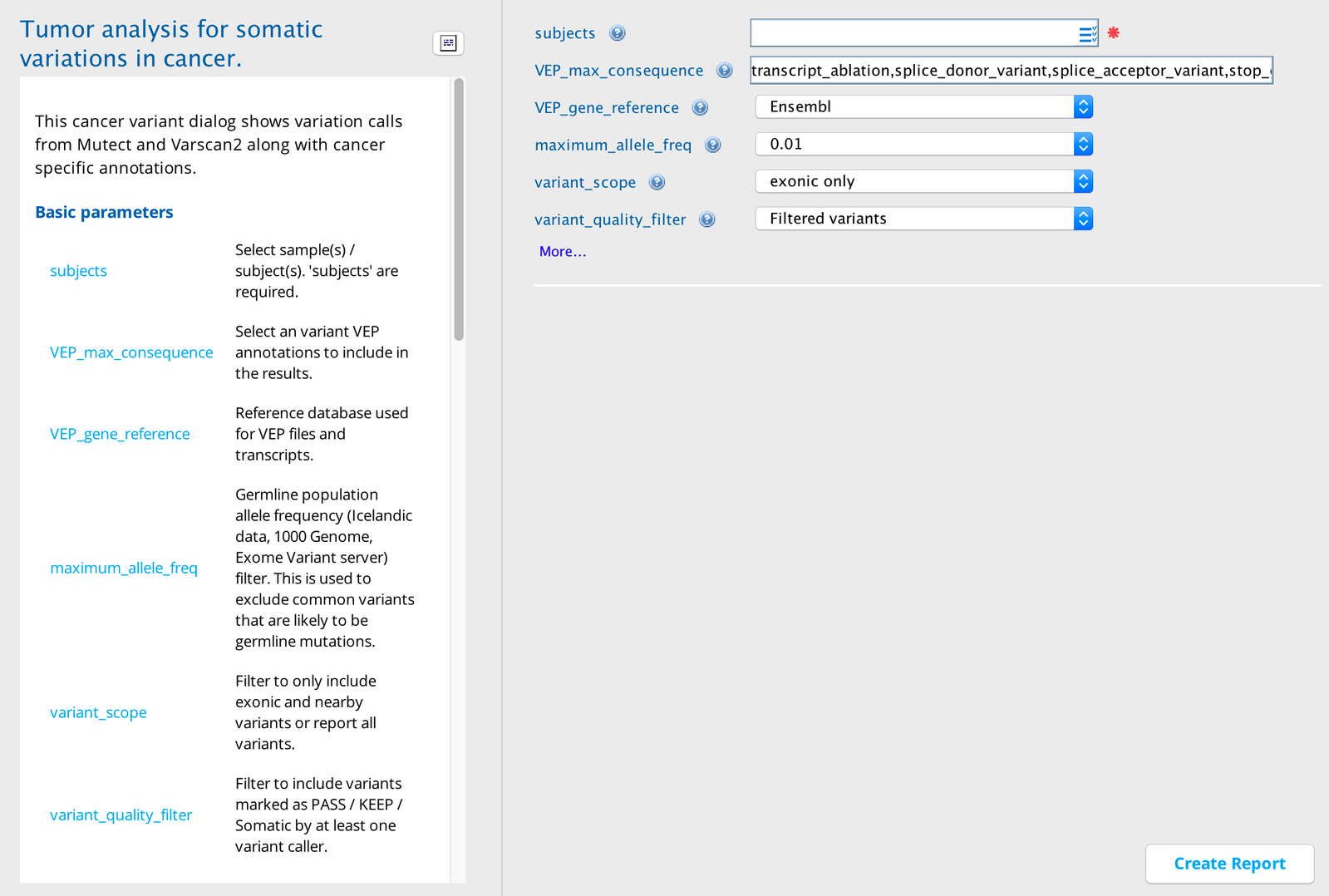
Tumor Mutation Analysis (TMA) in Sequence Miner¶
Example use case¶
The user has normal and tumor matched sample pairs and wishes to identify all likely somatic variants and to determine which variants are actionable and occur in known cancer genes.
Description of the algorithm¶
The processing pipeline utilizes the calls from each algorithm. The TMA applies user-defined filters to these lists of variants and adds several annotations.
Interpreting the output¶
The output can be displayed in Table view or Record view. The Table view of the output contains more than 90 columns. In general, these columns fall into three major categories:
Basic information (Chrom, Pos, Reference, Call, PN)
Annotations generated by the variant calling algorithms
Annotations from public data sources including COSMIC and GO annotations, actionable mutations and pathways, and clinical annotations
The Record view displays the variants in a list format and summarizes the most relevant information about the variant and key annotations:
Row 1: Chromosome position, gene, links to databases, transcript ID
Row 2: Nucleotide change, amino acid change and position, VEP maximum consequence, VEP maximum impact, maximum allele frequency, conservation score (maximum from SIFT and Polyphen-2)
Row 3: Callers for the variant, reads/depth in the tumor and germline samples
Row 4: COSMIC annotations

Record view¶
To toggle from Table view to Record view, click the list icon.
Column descriptions¶
Group |
Column |
Description |
|---|---|---|
Basic |
Call |
Sequence (variant) called, based on the reference sequence at the designated position |
Chrom |
Variant chromosome location |
|
PN |
Patient number (identifier) |
|
Pos |
Variant start bp position |
|
Reference |
Nucleotide from the reference build at the base pair position in the Pos column |
Group |
Column |
Description |
|---|---|---|
COMM |
CLINICAL_SIGNIFICANCE |
The clinical significance (e.g., pathogenic, benign, unknown significance, drug-response, risk factor, etc.) of the variant as annotated (commented) by users; if the same variant has several comments, this cell will contain a set of values |
MODE_OF_INHERITANCE |
The user-annotated (commented) mode of inheritance of the variant; if the same variant has been commented several times, this cell will contain a set of values |
|
TEXT |
The description (comment) component for the user annotation of the variant |
Group |
Column |
Description |
|---|---|---|
COSMIC |
count_lociInGene |
Count of COSMIC variants in this gene |
count_primarysitesInGene |
Number of primary site annotations in this gene |
|
count_vars |
Count of occurences of variants in COSMIC |
|
count_varsInGene |
Count of occurences of variants in this gene in COSMIC |
|
HGVSp |
Variant HGVSp notation using the preferred transcript; present only if the gene has a preferred transcript and the variant is found in this transcript |
|
IDs |
All COSMIC IDs for the variant |
|
primarysites |
List of primary site annotations for this variant |
|
primarysitesInGene |
List of primary site annotations in this gene |
|
pscount |
Number of primary site annotations for this variant |
Group |
Column |
Description |
|---|---|---|
Drug |
Gene |
Cancer drugs targeting this gene |
Gene_ApprovalStatus |
Approval status of the cancer drug |
|
Mechanism_of_Action |
Mechanism of action of the cancer drug |
|
Pathway |
Cancer drugs targeting a pathway of which this gene is a member |
|
Pathway_ApprovalStatus |
Approval status of the cancer drug |
|
SourcePathway |
The pathway on which the cancer drug acts |
Group |
Column |
Description |
|---|---|---|
Gene |
ActionableDrugClasses |
Drug classes associated with the gene encoding the variant |
ActionableMutations |
Clinically actionable mutations within genes |
|
ActionablePathways |
Clinically actionable pathways associated with genes |
|
ActionableTumors |
Clinically actionable tumors associated with genes |
|
Aliases |
List of gene aliases that correspond to the GENE_symbol |
|
listcount |
A count of the number of Gene_lists |
|
lists |
Gene panels or Gene lists |
|
Paralogs |
The paralogs of a given gene |
|
symbol |
Based on HGNC when it exists, otherwise it is the Ensembl internal alias |
Group |
Column |
Description |
|---|---|---|
Germ |
depth |
The number of reads used in evaluating the corresponding call |
depth_hq |
The number of high-quality reads used in evaluating the corresponding call |
|
reads |
The number of reads containing the variant call |
|
reads_hq |
The number of high quality reads containing the variant call |
Group |
Column |
Description |
|---|---|---|
GO |
Descriptions |
Gene Ontology category descriptions |
IDs |
Gene Ontology identifiers |
Group |
Column |
Description |
|---|---|---|
KNOWN |
dbSource |
Variants known in databases like HGMD, OMIM and ClinVar |
Gene_diseases |
Diseases known to be associated with the gene as annotated by HGMD, ClinVar, and OMIM |
|
lis_dbSourceMaxClinImpact |
A comma-delimited list of the KNOWN_dbsource and the known clinical impact of the gene (pathogenic/nonpathogenic, etc.) |
|
lis_disease |
A comma-delimited list of associated KNOWN_diseases |
|
MaxClinImpact |
The known clinical impact of the gene (pathogenic/nonpathogenic, etc.) |
|
var_diseases |
Diseases known to be associated with the variant as annotated by HGMD, ClinVar, and OMIM |
Group |
Column |
Description |
|---|---|---|
Mutect2 |
failure_reasons |
The reason MuTect judges a variant to be low confidence (REJECT) |
FORMAT |
Mutect2 Genotype Format Fields |
|
INFO: |
Mutect2 Genotype Format Info |
|
judgement |
MuTect judgement of site as somatic or not (KEEP or REJECT (not enough evidence or artifact)) |
|
NORMAL |
Genotype data from the normal sample |
|
Normal_LOD |
Log odds ratio (LOD) score for the normal sample |
|
TUMOR |
Log odds ratio (LOD) score for the normal sample |
|
Tumor_LOD |
Log odds ratio (LOD) score for tumor sample |
Group |
Column |
Description |
|---|---|---|
OMIM |
Descriptions |
OMIM disease descriptions for the gene |
IDs |
The OMIM ID of the gene |
Group |
Column |
Description |
|---|---|---|
Tumor |
High-quality reads are those with MAPQ >= 20 (MAPQ = -10*log10 (probability mapping position is incorrect)) |
|
depth_all |
The number of reads used in evaluating the corresponding call |
|
depth_hq |
The number of high-quality reads used in evaluating the corresponding call |
|
reads_all |
The number of reads containing the variant call |
|
reads_hq |
The number of high quality reads containing the variant call |
Group |
Column |
Description |
|---|---|---|
varscan2 |
FILTER |
Judgement of the variant |
FORMAT |
VarScan2 Genotype Format Fields |
|
HighConf |
VarScan2 further classify the somatic mutations as high-confidence or low-confidence |
|
INFO |
VarScan2 Genotype Format Info |
|
NORMAL |
Genotype data from the normal sample |
|
somatic_p_value |
Variant p-value for Somatic/LOH events |
|
somatic_p_score |
Score based on the p-value |
|
somatic_status |
Somatic status call (Germline, Somatic, LOH, or Unknown) |
|
TUMOR |
Genotype data from the tumor sample |
|
variant_p_value |
Variant p-value for germline events |
Group |
Column |
Description |
|---|---|---|
Variant |
Caller |
Variant caller used to judge the variant |
Caller_type |
Variant caller type: unpaired or paired |
|
PASS_count |
Number of variant callers that approve or “pass” the variant |
|
Present_count |
Number of variant callers that judge the variant |
Group |
Column |
Description |
|---|---|---|
Venn A |
In”Variant caller” |
A Boolean value if the variant has been rejected (0) or passed by the variant caller |
Group |
Column |
Description |
|---|---|---|
Venn B |
inGenelist |
A Boolean value if the variant is absent (0) or present (1) in a cancer gene list. |
WithPrimeSite |
Variant occurs in a tumor primary site as reported by COSMIC (1) or not (0) |
Group |
Column |
Description |
|---|---|---|
VEP |
Amino_Acids |
The amino acid change given as reference AA/variant AA; if the variation does not affect the protein-coding sequence, then “.” |
Biotype |
Biological class of transcript or regulatory feature |
|
CDS_position: |
Base pair position of the variant in the coding sequence; a value is given for each transcript |
|
HGVSp |
Variant notation with the transcript and amino acid change; there is an entry for each transcript |
|
HGVSp_simple |
Variant notation with the amino acid change; there is an entry for each transcript |
|
Max_Af |
Maximum reported allele frequency across the population surveys from 1000GP3, EVS, EXAC, Kyoto, GONL, and Iceland (Variant View-Frequencies) |
|
max_consequence |
Variant classes (high, moderate, low, and/or lowest impact on the gene product) |
|
Max_Impact |
Classification of the level of severity of the transcript consequence type assigned by VEP |
|
Max_Score |
Maximum score for the variant as observed in dbNSFP [Score=max ((1-Sift_score), Polyphen2_HDIV_score, Polyphen2_HVAR_score)] |
|
Protein_Position |
Position of the amino acid in the protein sequence (only if the variant falls within a coding sequence); a value is given for each corresponding transcript specified in the CDS position field |
|
Refgene |
The accession number from NCBI of the affected transcripts |
|
Transcript_count: |
Number of different transcripts in which the variant is found |
Group |
Column |
Description |
|---|---|---|
Other columns |
Annotation_Gene |
Cancer annotation for this gene |
CallRatio |
Proportion of reads containing the variant call; expected to be approximately 0.5 for heterozygous calls and close to 1 for homozygous calls |
|
candidate_gene |
Candidate genes entered by the user |
|
Category |
Denotes if variant is present in an oncogene or tumor suppressor |
|
dbNSFP_Interpro_domain |
The annotation from the dbNSF database, the domain of protein |
|
DIAG_ACMGcat |
Categorization of the sequence variants according to the ACMG scheme |
|
ReadDiff |
Difference in call ratio between tumor and normal samples |
|
Entrez-ID |
Entrez gene nomenclature |
|
ResistancexSensitivity_Variants |
Resistant to sensitivity information related to the variant |
|
SubjectId |
Patient ID |
Perspective views¶
The Default view perspective lists all variants depending on the variantFilter criteria selected in the input parameters. Additional perspectives focus on subsets of the columns in the default view.
Perspective |
Description |
|---|---|
Candidate genes |
Somatic variants present in the user’s candidate genes report file (if selected from the user in the input parameters) |
Default view |
Lists all variants depending on the variantFilter criteria selected in the input parameters, e.g., if the user selects “Filtered variants”, this perspective will list the same number of variants shown in the “Filtered” perspective |
Filtered |
Somatic variants that meet the quality criteria (present in at least 2 high-quality reads and ratio of hq reads to depth in tumor is greater than 0.01); high-quality reads are those with MAPQ ≥ 20 (MAPQ = -10*log10 (probability mapping position is incorrect)) |
Filtered cancer genes |
Somatic variants that meet the quality criteria (see Filtered perspective above) and are present in cancer-associated genes (from commercial gene panels); the list of commercial gene panels is listed in the |
Filtered candidate genes |
Somatic variants that meet the quality criteria (see Filtered perspective above) and are present in the user’s candidate genes report file (if selected from the user in the input parameters) |
The user can then filter on additional annotation (e.g., COSMIC annotation, known associated gene disease, and actionable mutations/pathways) to further narrow the list of candidate genes and gene variants. These annotations can be found in the right-hand pane, which lists all of the columns, grouped by category.