Regression and SKAT analyses¶
Sequence Miner is a research tool for integrated analysis of genotype and phenotype data on patients. Clinical and other phenotype data can be imported into Sequence Miner through the metadata query language NOR (Non-Ordered Relational). Data fields can be sorted and filtered allowing the user to quickly classify patients into categories (e.g., affected and unaffected) The NOR-defined subsets can then be used as input list(s) for genomic analysis applications in Sequence Miner called report builders that apply the GOR (Genomically Ordered Relational) query language.
Sequence Miner has two report builders for analyzing covariate data in the context of genomic variants: Single point regression and Sequence Kernel Association Test (SKAT). The Single point regression report builder performs linear or logistic regression analysis to measure the relationship between a given outcome variable (e.g., diagnosis) and covariates in the context of genomic variants in the cases and controls. The SKAT report builder measures the relationship between covariates and the distribution of genomic variants in the cases and controls.
Both analyses require a Phenogrid as input. A Phenogrid lists samples with an outcome variable (e.g., case, control) followed by values for any number of covariates. The covariates may be continuous or dichotomous.
This guide outlines the following workflow to conduct Single point regression and SKAT analyses in Sequence Miner:
Preparing a Phenogrid containing numerical outcome and covariate values for selected subjects
Running the Single point regression or SKAT report builder
Interpreting the results
This workflow is shown in more detail below:

Regression and SKAT analysis workflow¶
Preparing a Phenogrid¶
The first step is define the case and control (outcome) groups. Cases and controls can be selected by filtering on features such as an identification in the sample name or a qualitative/quantitative attribute. These features can be found in a user-generated phenotype data file, created from a grid containing phenotypes and/or clinical data for individuals with corresponding genomic data.
To access this file, open Sequence Miner from the CSA dashboard. In the Sequence Miner File Explorer, select the desired file and double-click the file name to open it in a new tab. This metadata file must include the list of sample IDs as the leftmost column, and the column should be named “PN”. For the filtered “case” or “control” lists to be used for querying the genotypic data, the sample IDs in the PN column must be identical to those assigned to the genotype files (e.g., BAM and VCF). A phenotype data file also contains columns for the phenotype covariates (gender, ethnicity, BMI, weight, age, etc.).

Sort the columns by clicking on the column header. A triangle appears next to the selected column to indicate ascending order (triangle pointing up) or descending order (triangle pointing down). Click the triangle to toggle the sort order. To filter on a column, right-click on the column name and select Filter on Column.
Select the desired subjects (e.g., a case group) by typing in a filter term. Select the check boxes at the top and bottom of the filtered list, and then right-click and select Set Selected. Click Apply to view only the selected columns in the table.

Columns can also be filtered when numerical values are present. To define the PN (patient) set, right-click the column name and select the desired cutoff value. Filtered columns appear in green at the top of the Columns panel on the right-hand side.

Create a “case” or “control” group from the filtered list by highlighting the PN column and right-clicking on the highlighted cells, including the phenotype columns as shown. Select Open in new Grid as Cases or Open in new Grid as Controls. The new grid with the PN list, labeled “Cases” or “Controls” depending on the selection made, opens in a new Sequence Miner window.

Open the Phenogrid report builder from the Report Builders tab. You can select CaseControl in the Category filter to filter only Case/Control Reports, and then click on the Phenogrid report builder in the menu grid.

Select the grid for CASEs and CTRLs by clicking inside the fields in the Phenogrid report builder dialog and selecting the file from the drop-down list. The files must be open in the browser in order to select them as inputs. Click Create Report.
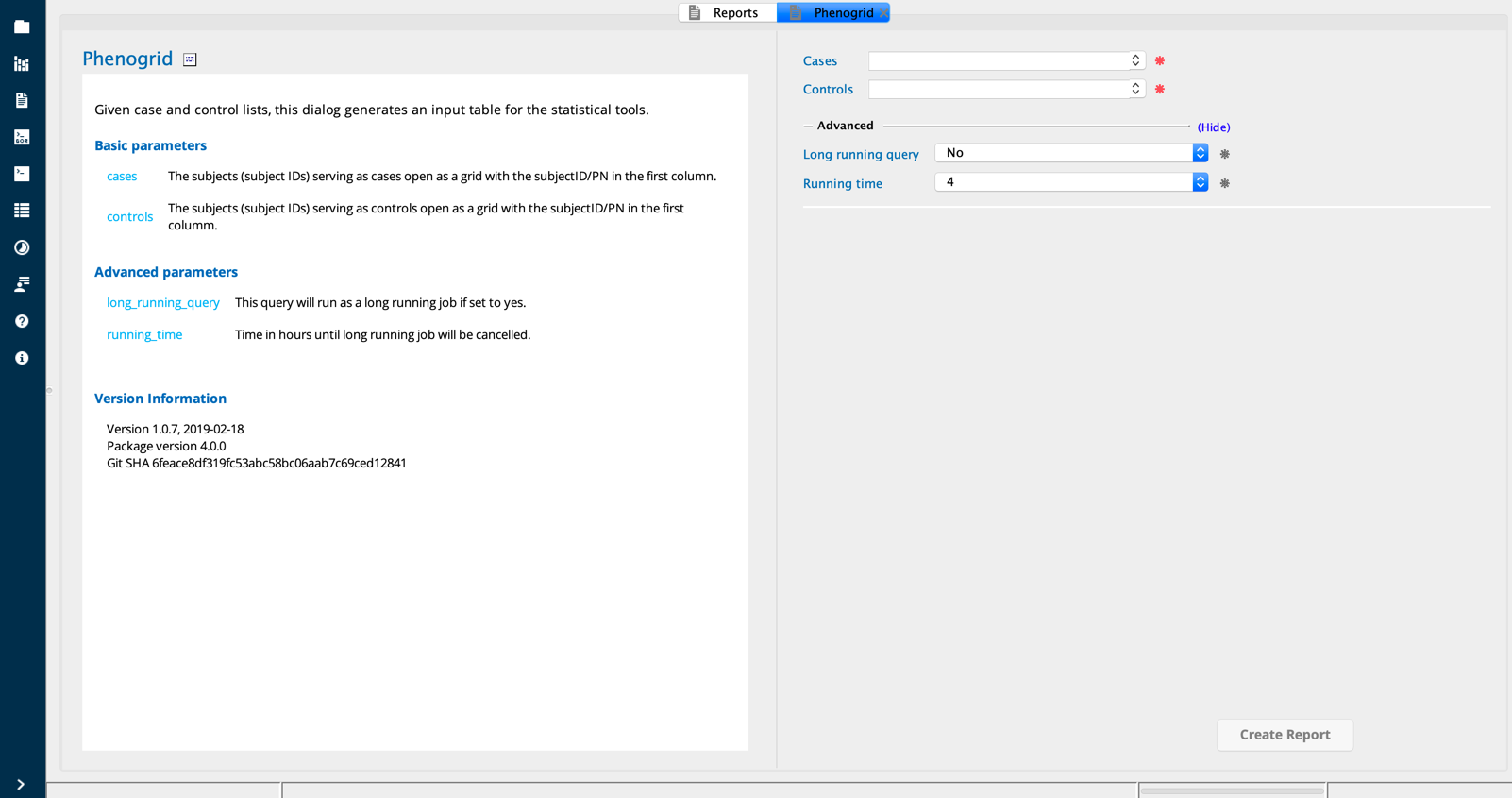
The Phenogrid report builder combines the two case and control grids into a single grid with a new Affected column in which cases have a value of “1” and controls have a value of “0”. This column is used as the outcome measure for downstream analysis.
Note
The Affected column contains binary values. This format can be used for logistic Single point regression and the dichotomous mode for SKAT.

All phenotypic values must be numerical. Therefore, for compatability with downstream analysis tools, all columns that contain string values must be converted to numerical values.
Additionally, all categorical values must be transformed into dichotomous values. For cases in which there are more than two states for a categorical value (e.g., ethnicity), each categorical state must be given a separate column. Each sample is then designated as either exhibiting that categorical state (“1”) or not (“0”).
For example, the Gender column in the table shown below contains text values for “male” and “female” that must be converted to “0” and “1”. Likewise, the Ethnicity column contains text and categorical values. The text values should be converted to “0” and “1” for each listed ethnicity in separate columns. For instance, the Ethnicity column contains four values: “Caucasian”, “AfrAmer”, “AsianAmer”, and “Hispanic”; this column will be split into four columns with “0” and “1” values for each ethnicity.
To convert text values and/or categorical values, click the Add Calculated Column icon in the toolbar above the grid.

In the dialog box that opens, type a new column identifier in the Name field. Note the following when entering column names:
Names must begin with a text value (column names may not begin with a numerical value or special characters).
Names may not contain spaces or special characters (use “_” instead of a space).
Columns that contain a similar prefix will be grouped together (e.g., columns named “ethnicity_Cauc” and “ethnicity_AfrAmer” will be grouped as “ethnicity”).
To define a Boolean to convert the columns, click Boolean.

An “if” statement is introduced. In the following example, the text column Gender (shown in blue) is converted to the column PN_female (shown in the Name field) where the string value “female” is converted to “1” and “male” to “0”. Similarly, the Ethnicity value “Caucasian” is converted to a new column called PN_Caucasian containing “1” for Caucasian and “0” for non-Caucasian.

The original table expands to include the added columns for gender and ethnicity.

The original columns can be hidden by either deselecting the column in the Columns panel or right-clicking the column name and selecting Hide Column.

The final Phenogrid shows only numerical values, as in the example below.

Running the Single Point Regression report builder¶
After defining the case and control groups, the next step is to identify the appropriate report builder. Report builders are categorized according to the type of analysis they perform.
To view the options for analysis, open the Reports tab. Select CaseControl in the Category filter to see the report builders that accept “case” and “control” grids for statistical analysis. Open the Single point regression report builder by clicking on it in the menu grid.

Select as input the Phenogrid file you created in the first step. The regression tool requires as input at least two columns in the Phenogrid file: PN (first column) and outcome (second column).
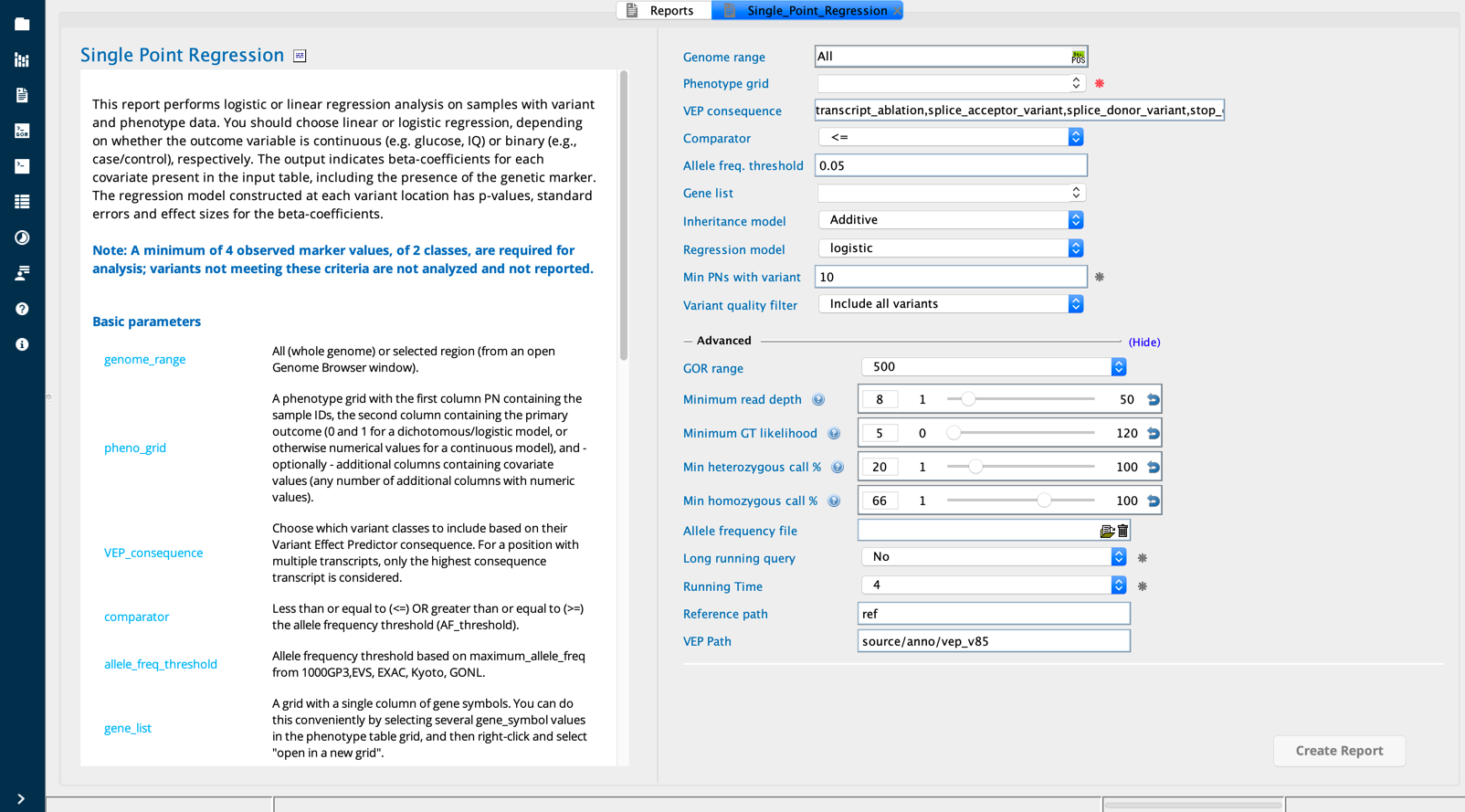
The Single point regression report builder performs single-point linear or logistic regression analysis on samples with sequence variants and covariates.
Regression analysis determines the relationship between an explanatory variable (the independent) and the outcome, the response variable (the dependent variable, y). While linear regression fits continuous response data (e.g., tumor reduction, BMI, blood pressure) to a linear data model, logistic regression is used to model dichotomous response data (affected/unaffected). This analysis requires a minimum of four variant carriers (het or hom) and two homozygous for the reference call.
A Phenogrid is the only required input. In the Regression field, select the regression type (linear or logistic regression). In the Model field, select the model (additive, autosomal recesive, or autosomal dominant). For the remaining filters, you can accept the default settings or select filters for the following:
genome_range
variant scope
allele frequency threshold
VEP_consequence
minimum PNs with the variant, variant filter, minimum genotype (GT) quality, minimum het call percent, minimum hom call percent, and minimum read depth
These filters are applied to the variant data on each sample in the analysis as follows:
Variants are selected for the analysis in the query:
All variants in coding sequence (+/- 1000bp) from the whole exome sequence data in the project are filtered by the user-designated thresholds for VEP_max_consequence and allele frequency threshold.
Of these, only the variants that are also carried by at least the min_PNs_with_var (minimum number of samples) designated by the user are kept for the next filtering steps.
Next, the query calculates a quality value for each variant based on the user-designated thresholds for variant_filter (PASS or low quality), min_GT_quality, min_read_depth, and min_call_perc.
The remaining variants are filtered based on that quality value plus the min_read_depth for the samples carrying the variant.
Finally, the variant is assigned a value based on zygosity and coverage at that position.
If a variant has segment coverage less than 8X, the variant is assigned “NA”.
If the variant has good coverage (at least 8X segment coverage) and is heterozygous, the variant is assigned a value of “1”.
If the variant has good coverage (at least 8X segment coverage) and is homozygous, the variant is assigned a value of “2”.
If the variant has good coverage (at least 8X segment coverage) and is not heterozygous or homozygous, the variant is assigned a value of “0”(homozygous reference).
When the analysis is complete, a new table window opens in Sequence Miner. For each variant, this query returns statistical measures for each covariate, one row per covariate. These statistical measures include:
goodness of fit (GOF)
b-coefficient
standard error
standard score (Z-score)
p-value
effect size
alpha value (corrected p-value)
Firth correction statistic (bias correction for data separation)

Sort columns by clicking the column header. To filter columns, right-click the column name. Filtering can be used to define cutoffs for different numerical values based on the distribution of the column data or by free text searching for string values.

Variants of interest can be further annotated with a drill-in report To view available drill-in report annotations, right-click on the variant row(s) of interest. Select Drill in Reports and choose from VEP, dbNSFP, dbSNP, HGMD, ClinVar, 1000G, or EVS annotations.

Once a drill-in report has been selected, a new window opens that shows the listed variant(s) and/or gene(s) with the selected annotations.

Interpreting the regression analysis results¶
Column |
Description |
|---|---|
GOF |
Goodness of fit of the model for the given set of covariates (correlation value from 0- 1, where “1” is the best fit) |
name |
Names of all explanatory variables (x) in the regression model |
coeff |
b coefficient is the standardized weight of the predictor in the model (higher the value, the greater the contribution to the model); b coefficient (b coefficient and effectSize are the same for linear regression) |
stdError |
Standard error to measure the precision of the estimate of the b coefficient (σ2/√N, where N = sample size) |
stdScore |
Standard score is the Z-score (the number of standard deviations away from the mean of the sample distribution: b coeff / stderror) |
pValue |
The p-value (the probability of obtaining a result at least as extreme as the observed result) |
effectSize |
|
Firth |
Bias correction statistic for data separation |
Note
A covariate with a p-value of 2.0 indicates that a covariate has invariant data (a single value for all samples. In this case, the analysis should be rerun after removing (hiding) the invariant covariate column in the Phenogrid.
Analyzing the regression analysis results¶
The p-value column may first be sorted to view the covariates with the most statistically significant correlation to the outcome.
Next, these p-values are evaluated against the alpha column, which contains the Bonferroni-corrected statistical significance threshold (e.g., based on a statistical significance threshold of 0.05):
alpha = 0.05 / (total # of markers)
P-values of less than alpha indicate statistical significance of the covariate to the outcome.
Covariates that meet this criteria are further evaluated based on the effectSize, which reflects the contribution of each covariate to the outcome.
The GOF (goodness of fit value) reflects the effectSize of the whole model: the combined contributions of the covariates plus the variant to the outcome.
Running a SKAT analysis with report builders¶
The Sequence Kernel Association Test (SKAT) is a variance-component test for genetic association analysis. SKAT is a SNP-set level test that evaluates the distribution of genetic effects (both positive and negative) of a group of variants on continuous or dichotomous traits. Instead of aggregating variants into an aggregate-allele (e.g., the Gene analysis report builder), SKAT evaluates the distribution of the aggregated score statistics of individual variants involved in continuous or dichotomous traits. This tool incorporates the SKAT-O R statistical package.
A Phenogrid is the only required input. The Phenogrid must have at least two columns: PN (first column) and outcome (second column). Additional covariates may be added in columns 3-n. Select as input the Phenogrid file you created in the first step.
To view the options for analysis, open the Reports tab. Select CaseControl in the Category filter to see the report builders that accept “case” and “control” grids for statistical analysis. Open the SKAT report builder by clicking on it in the menu grid.

In the ResponseVariableType field, select the appropriate setting to designate whether the response variable (the second column in the Phenogrid) is dichotomous (“D”, e.g., affected or unaffected) or continuous (“C”, e.g., BMI). In the Covariates field, select a setting for covariates (covariates/no covariates). For the remaining filters, you can accept the default settings or select filters for the following:
genome_range
variant scope
allele frequency threshold
VEP_consequence
variant filter
minimum genotype (GT) quality
minimum het call percent
minimum hom call percent
minimum PNs with the variant
minimum read depth
Variants are selected for the analysis in the query as follows:
All variants in coding sequence (+/- 1000bp) from the whole exome sequence data in the project are filtered by the user-designated thresholds for VEP_max_consequence and allele frequency threshold.
Of these, only the variants that are also carried by at least the min_PNs_with_var (minimum number of samples) designated by the user are kept for the next filtering steps.
Next, the query calculates a quality value for each variant based on the user-designated thresholds for variant_filter (PASS or low quality), min_GT_quality, min_read_depth, and min_call_perc.
The remaining variants are filtered based on that quality value plus the min_read_depth for the samples carrying the variant.
Finally, the variant is assigned a value based on zygosity and coverage at that position:
If a variant has segment coverage less than 8X, the variant is assigned “NA”.
If the variant has good coverage (at least 8X segment coverage) and is heterozygous, the variant is assigned a value of “1”.
If the variant has good coverage (at least 8X segment coverage) and is homozygous, the variant is assigned a value of “2”.
If the variant has good coverage (at least 8X segment coverage) and is not heterozygous or homozygous, the variant is assigned a value of “0” (homozygous reference).
The analysis requires that no more than 20% of the samples are missing a given variant.
When the analysis is complete, a new table window opens in Sequence Miner. For each gene, this query returns a SKAT p-value for the association between the gene and the response variable (in the context of the covariates, if applicable).

Sort columns by clicking the column header. To filter columns, right-click the column name. Filtering can be used to define cutoffs for different numerical values based on the distribution of the p-values or by free text searching for string values.

Variants of interest can be further annotated with a drill-in report. To view available drill-in report annotations, right-click the variant row(s) of interest, select Drill in Reports, and choose from gene Pathways and gene Clinical disease gene annotations. This opens a pop-up window with the listed genes and selected annotations.

Interpreting the SKAT analysis results¶
Column |
Description |
|---|---|
Chrom |
The chromosome of the region |
Gene_start |
The start base pair position of the gene (zero based, i.e., the position of the base pair before the first base pair in the gene) |
Gene_end |
The end base pair position of the gene |
skat_pvlue |
p-value representing the probability that the observed distribution of variants in the given gene across cases, controls, and covariates (if applicable) occurred by chance |
Note
A p-value of “1.0” indicates insufficient information to perform the analysis for that gene (e.g., more than 20% of the samples are missing the variant, a covariate is invariant, or there are missing values in the covariates).