Gene analysis¶
Description of the report¶
The Gene analysis report builder provides a summary count of the variants identified in genes for a defined set of individuals (cases) versus the count for the same variants in all other individuals in the project. The summary counts are generated for the individual variant and for alternative variants within the same gene. This includes a count of individuals homozygous for any alternative variant in the gene.
By default, the statistics are calculated for the Ensembl reference gene set. Alternatively, the results may be restricted to a user-defined gene list.
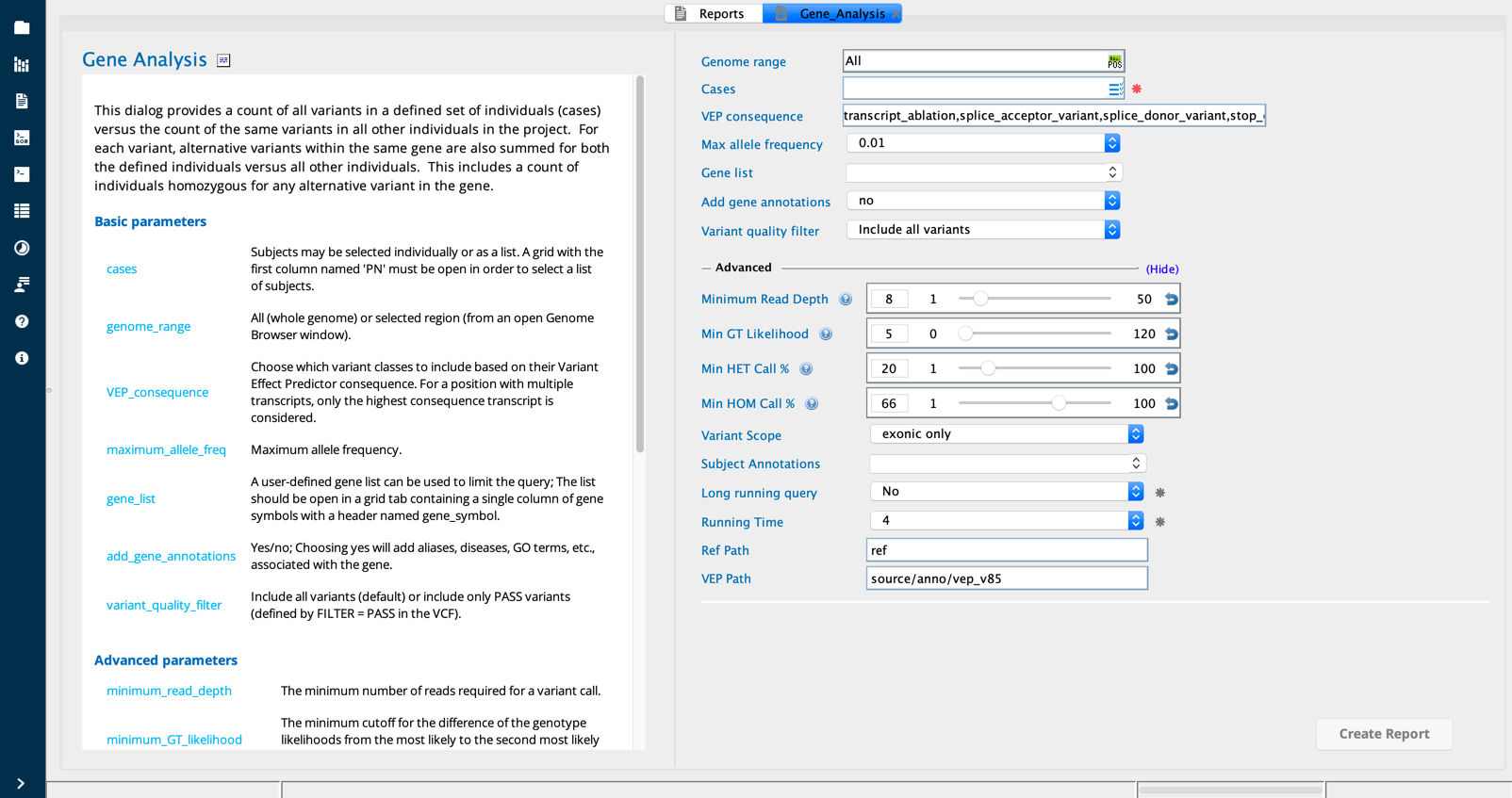
Gene analysis in Sequence Miner¶
Example use case¶
For each gene in a given gene-list, the user may wish to do the following:
Identify unique loss-of-function variants in the “case” group, which are not found in other individuals in the project
Find genes with a higher occurrence of truncating variants in the case group compared to all other samples in the project
As an example, several subjects in the case group may be homozygous for a truncating variant and the user may wish to tally the number of other subjects in the project who carry this variant, are homozygous for the variant, and carry alternative variants in the same gene.
This report builder can also add clinical annotation to every variant in the report.
Description of the algorithm¶
All variants in the project are stored in a central repository on the server along with sample source and data quality information. This report builder query retrieves all variants for a given set of samples and optionally filters out low-quality, common, and low-impact variants. The filtered variants are tallied to generate a count of the number of samples (designated as “CASEs” for selected samples and “OTHERs” for all other subjects in the project) containing the same variant.
Additionally, these variants are mapped onto an Ensembl reference set of genes to tally the number of samples (“CASEs” or “OTHERs”) that share variants in the same gene. VEP predicted-functional-effect annotation is added to the variants and genes.
Interpreting the output¶
The resulting table tallies the counts of variants and genes with variants for the CASEs and OTHERs groups. These columns can be used to compare, for example, the number of cases homozygous for a truncating variant (using the CASEs_hom and VEP_Max_Impact columns) compared to the remaining samples in the project (using the OTHERs_hom column).
There are several columns to consider for an overview of the analysis results.
Column descriptions¶
Group |
Column |
Description |
|---|---|---|
Basic |
Call |
|
Chrom |
||
POS |
||
Reference |
||
CASEs |
hom |
The number of designated cases homozygous for the variant |
hom_InGene |
The number of designated cases with a homozygous variant in the indicated gene |
|
var |
The number of designated cases containing the variant (includes heterozygous and homozygous) |
|
var_InGene |
The number of designated cases with a variant in the gene (includes heterozygous and homozygous) |
|
OTHERs |
hom |
The number of all other subjects (subtract selected cases from the subjects in the project) homozygous for this variant |
hom_InGene |
The aggregated number of all other subjects (subtract selected cases from the subjects in the project) with a homozygous variant in the gene |
|
var |
The number of other subjects (subtract selected cases from the subjects in the project) containing this variant (includes heterozygous and homozygous) |
|
var_InGene |
The aggregated number of variants from all other subjects (subtract selected cases from the subjects in the project) in the gene |
|
VEP |
Amino_Acids |
The amino acid with and without variant (only provided if the variant affects the protein-coding sequence), otherwise “.” |
CDS_position |
Position of the base pair in the coding sequence; a value is given for each transcript |
|
max_af |
Maximum reported allele frequency across the population surveys from 1000GP3, EVS, EXAC, Kyoto, GONL |
|
max_consequence |
Classification of the level of severity of the transcript consequence type assigned by VEP |
|
Max_Impact |
VEP predicted consequence for a variant producing the the greatest impact on the transcript |
|
Protein_Position |
Position of the amino acid in the protein sequence (only if the variant falls within a coding sequence); a value is given for each corresponding transcipt specified in the CDS position field |
|
Refgene |
The accession number from NCBI of the affected transcripts |
|
Other columns |
GENE_SYMBOL |
Based on HGNC when it exists, otherwise it is the Ensembl internal alias |
The user may compare the CASEs_var and OTHERs_var columns first to evaluate how common a specific identified variant is among cases.
The user may focus on CASEs_hom and OTHERs_hom if interested in homozygous carriers, or CASEs_var_InGene and OTHERs_var_InGene for compound heterozygous carriers.
Perspective views¶
The Default view perspective lists all variants identified in the CASEs. Additional Perspectives subtabs focus on subsets of columns in the default view.
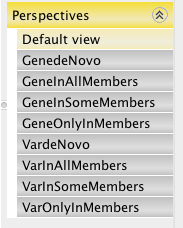
Perspectives in the Gene analysis report builder¶
Perspective |
Description |
|---|---|
Default view |
Lists all the variants identified in the CASEs. |
GenedeNovo |
Lists genes (by variant) that contain one variant in the CASEs group but do not contain variants from all other subjects (CASEs_var_InGene = 1 and OTHERs_var_InGene = 0). |
GeneInAllMembers |
Lists genes (by variant) in which all cases share a variant (CASEs_var_InGene = total number of selected samples). |
GeneInSomeMembers |
Lists genes (by variant) in which any of the cases selected contains a variant (CASEs_var_InGene > 0). |
GeneOnlyInMembers |
Lists genes (by variant) uniquely containing variants from the cases but not any other subjects in the project (CASEs_var_InGene >= 1 and OTHERs_var_InGene = 0) |
VardeNovo |
List variants that are uniquely present in one of the selected case (CASEs_var = 1 and OTHERs_var = 0). |
VarInAllMembers |
List variants that exist in all the cases selected (CASEs_var = total number of selected samples). |
VarInSomeMembers |
List variants that exist in any one of the cases (CASEs_var > 0). |
VarOnlyInMembers |
List variants that exist uniquely in selected cases but not in other samples (CASEs_var >= 1 and OTHERs_var = 0). |