Venn variant analysis¶
The Venn variant analysis dialog reports overlap between the variants in two to four GOR reports at the variant, gene, or pathway level. A Venn diagram can be generated in the completed report to visualize the fraction of variants, genes, or pathways that overlap between the selected GOR reports.
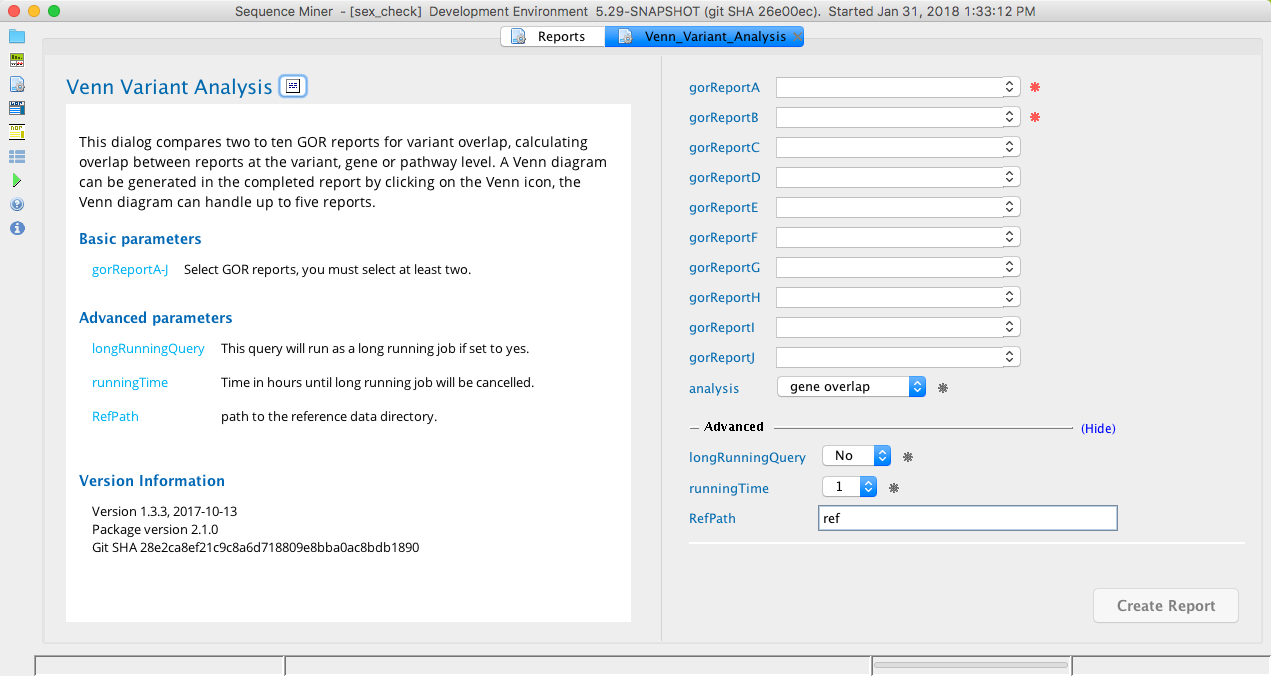
Venn Variant Analysis module in Sequence Miner¶
Example use case¶
A user wishes to identify variants common to two possibly genetically-related phenotypes and runs the Inheritance report builder separately on two sets of cases:
Cases with progressive vision loss
Cases with progressive hearing loss
The user can run this report builder using each Inheritance report as input to identify variants common to both sets of cases and those unique to one set or the other. Any variant report with the following four columns can be used as input for this report builder: chrom, pos, reference, call.
Interpreting the output¶
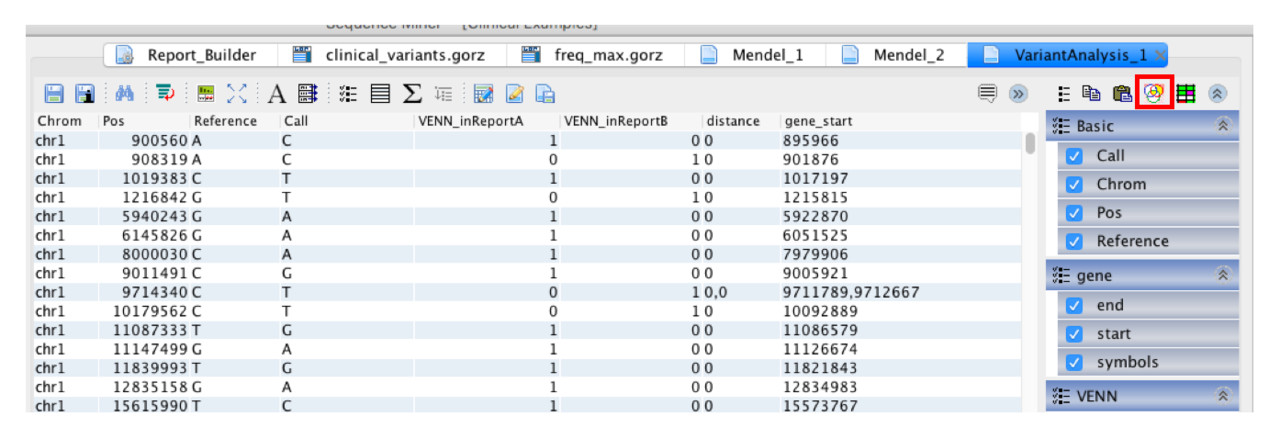
Step 1: Select two to four GOR reports to compare and click Create Report. The VENN_inReportA/VENN_inReportB column indicates whether a variant is in a report or not. The Venn tool uses only the columns with a “1” or “0” to indicate the presence or absence of a shared feature (variant, gene, or pathway) in a variant report.
Step 2: Click the Show Venn Filter icon.
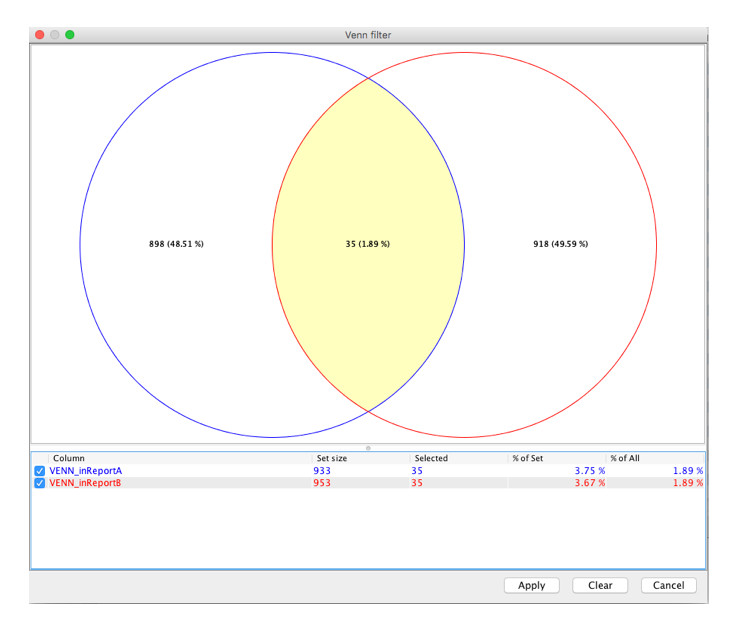
Step 3: Select a region on the Venn diagram and click Apply to apply the filter to the table.
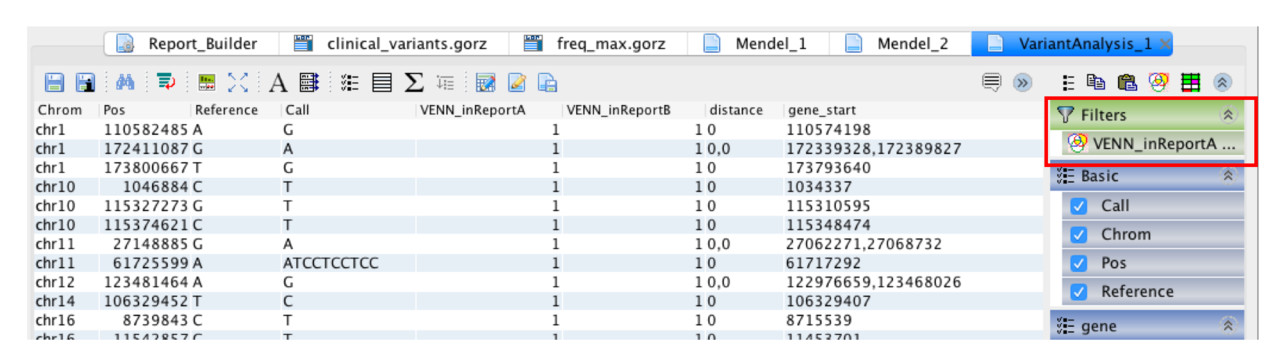
Step 4: Display only the variants selected in the Venn diagram.
Column descriptions¶
Group |
Column |
Description |
|---|---|---|
gene |
end |
The end base pair position of the gene |
start |
The start base pair position of the gene (zero-based, i.e., the position of the base pair before the first base pair in the gene) |
|
Symbol |
Based on HGNC when it exists, otherwise it is the Ensembl internal alias |
|
VENN |
inReportA |
A Boolean column (0/1) indicating whether a variant is in a report or not |
inReportB |
A Boolean column (0/1) indicating whether a variant is in a report or not |
|
Other columns |
Chrom |
The chromosome of the variant, represented as chr1, chr2, …, chr22, chrXY, chrX, chrY, chrM |
distance |
In the case where the user selects the variant overlap or gene overlap options, a fuzz factor is applied to include nearby genes up to 20 bp’s away from the variant that may be affected by the variant. The output from the variant overlap option includes an additional column called “distance” which shows the number of base pairs between the variant and a nearby gene lying within 20 base pairs. |