Variant count¶
The Variant count report builder summarizes the variants and their zygosity found in a group of user-defined affected subjects (cases) versus unaffected subjects (controls). The summary includes a tally (for cases and controls) of variants at the gene level plus hom and het tallies and fractions at the variant level.
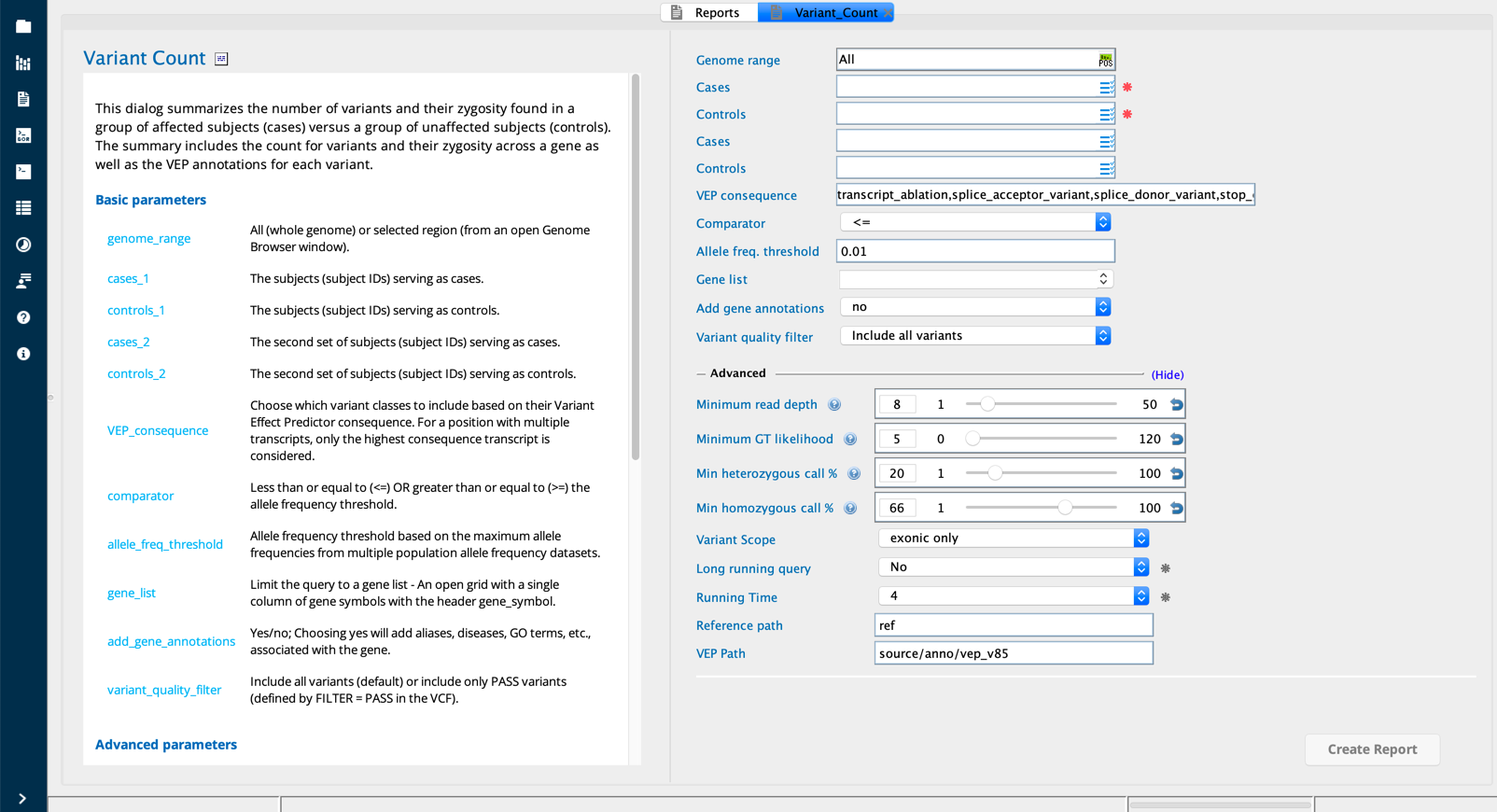
Variant Count module in Sequence Miner¶
Example use case¶
A researcher has a cohort of subjects with (cases) and without (controls) autism. She wishes to compare the fraction of homozygous variants in the cases versus the controls.
Conducting a variant level analysis¶
Compare the ratio columns (for het or hom carriers) in cases versus controls. The user can, for example, add a calculated (e.g., Boolean) column to highlight those variants carried by a greater proportion of het/hom cases compared to het/hom controls:
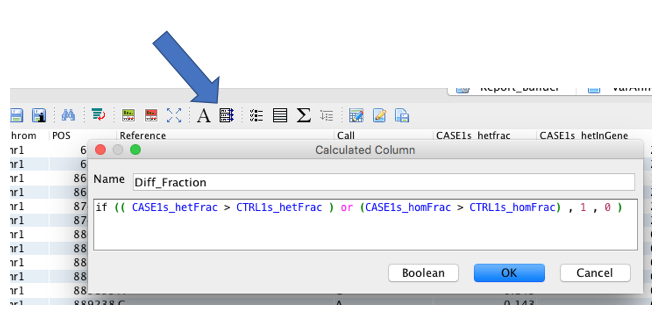
if (( CASE1s_hetFrac > CTRL1s_hetFrac ) or (CASE1s_homFrac > CTRL1s_homFrac) , 1 , 0 )¶
Conducting a gene level analysis¶
The analysis returns only one variant per row. The hetInGene and homInGene columns for cases and controls tally the total variant load for each genotype (het or hom) per gene. For each genotype (het or hom), compare the values in these columns between cases and controls to identify differences in variant load per gene.
Description of the algorithm¶
This query first filters variants meeting the user-defined VEP consequence and user-defined quality filters. Next, the number of heterozygous and homozygous cases and controls are tallied for every variant. A count-fraction for heterozygous and homozygous cases and controls is also returned for every variant allele. The analysis also returns hetInGene and homInGene columns, which display the total variant load for each genotype (het or hom) per gene.
Interpreting the output¶
This report builder lists one variant per row. The results are returned for cases and controls separately and fall into three categories:
Per variant ratio (fraction)
Per variant carriers
Carriers of any variant per gene
Column descriptions¶
Group |
Column |
Description |
|---|---|---|
Basic |
Call |
|
Chrom |
||
POS |
||
Reference |
||
cases |
1_hetfrac |
The ratio between cases heterozygous for the variant and total number of selected cases; cases_1_hetInVar/(total cases) |
1_hetInGene |
The number of cases that contain heterozygous variants in the gene mapped to the variant position |
|
1_hetInVar |
The number of cases with this variant in a heterozygous state |
|
1_homfrac |
The ratio between cases homozygous for the variant and total number of selected cases; cases_1_homInVar/(total cases) |
|
1_homInGene |
The number of cases that have homozygous variants in the gene mapped to the variant position |
|
1_homInVar |
The number of cases with this variant in a homozygous state |
|
controls |
1_hetfrac |
The ratio between controls heterozygous for the variant and total number of selected controls; controls_1_hetInVar/(total controls) |
1_hetInGene |
The number of controls that contain heterozygous variants in the gene mapped to the variant position |
|
1_hetInVar |
The number of controls with this variant in a heterozygous state |
|
1_homfrac |
The ratio between controls homozygous for the variant and total number of controls; controls_1_homInVar/(total cases) |
|
1_homInGene |
The number of controls that have homozygous variants in the gene mapped to the variant position |
|
1_homInVar |
The number of controls with this variant in a homozygous state |
|
VEP |
CDS_position |
Position of the base pair in the coding sequence; a value is given for each transcript |
max_af |
Maximum allele frequency from public databases (1000Genomes, Exome Variant Server, ExAC, GONL, Kyoto) |
|
max_consequence |
Variant classes (high, moderate, low, and/or lowest impact on the gene product) |
|
Max_Impact |
Classification of the level of severity of the transcript consequence type assigned by VEP |
|
Refgene |
The Accession number from NCBI of the affected transcripts |
|
Other columns |
Gene_Symbol |
Based on HGNC when it exists, otherwise it is the Ensembl internal alias |