Transcripts¶
The Transcripts report builder comprehensively annotates a given variant to assist the user in evaluating the effect of the variant. Annotation includes consequence and max consequence reported for the variant, based on transcripts drawn from the source/anno/vep_3-4-2/vep_multi_wgs.gord file (in the Sequence Miner File Explorer). The vep_multi_wgs.gord file lists all reported transcripts for the following:
A single variant at a given locus
Multiple variants at the same locus
Nearby and overlapping upstream and downstream genes/pseudogenes
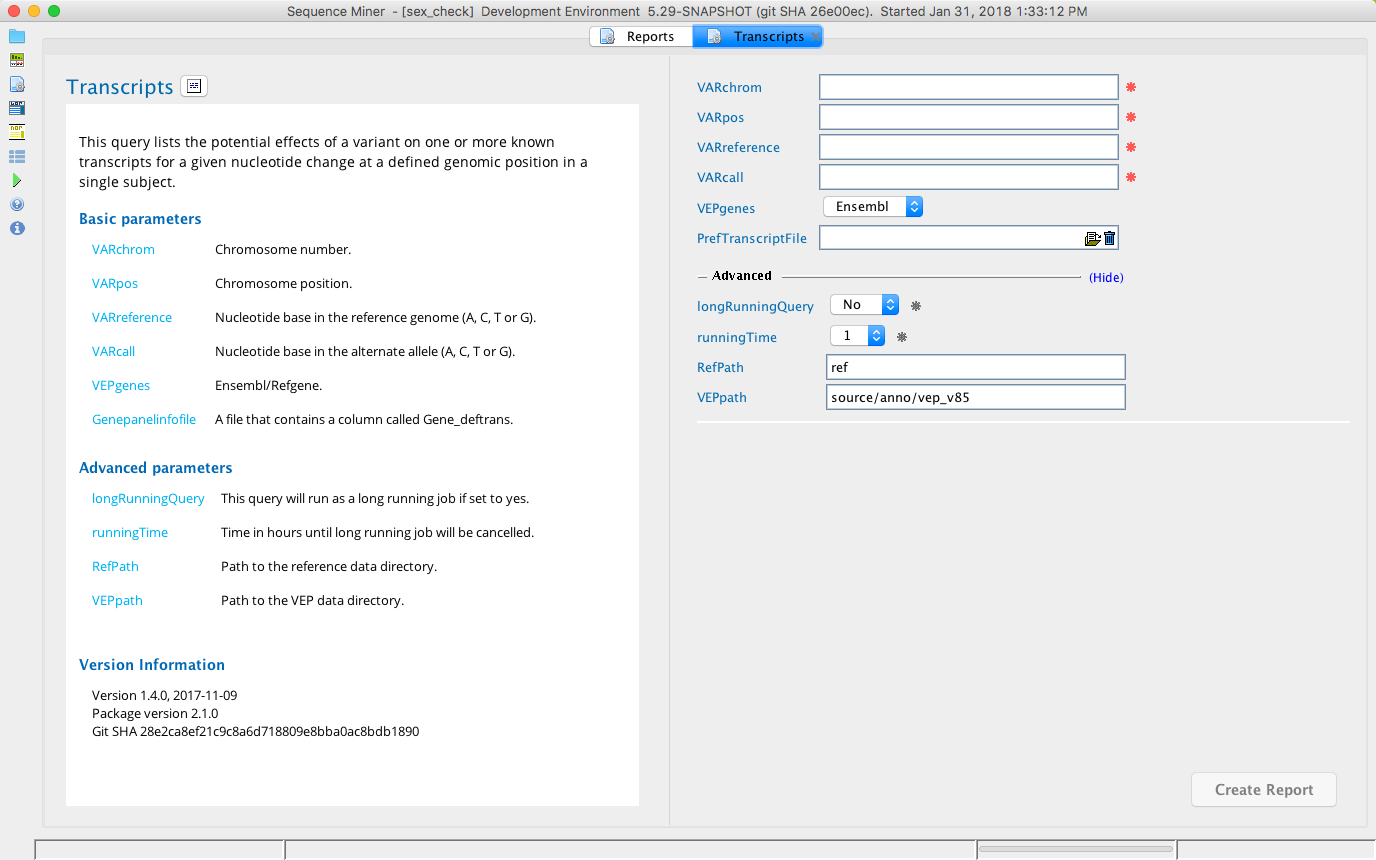
Transcripts module in Sequence Miner¶
Example use case¶
The user has identified a variant in a case and wishes to annotate the variant with all known transcripts along with the associated variant effect consequences and the maximum observed consequence among all transcripts carrying the variant.
Description of the algorithm¶
This query annotates the input variant with structural and functional features of the sequence (e.g., protein domain information). The annotation also includes IDs for transcripts carrying the variant or near the variant (in these cases, the distance column lists the number of bases between the variant and the transcript) along with the associated transcript-annotation from the Variant Effect Predictor (VEP) algorithm.
Interpreting the output¶
Column descriptions¶
Group |
Column |
Description |
|---|---|---|
Basic |
Call |
The actual called sequence (variant), found by replacing a part of the reference sequence, denoted by Pos and Reference, with the sequence in the Call column |
chrom |
The chromosome of the variant, represented as chr1, chr2, …, chr22, chrXY, chrX, chrY, chrM |
|
Pos |
The (first) base pair position of the sequence variant, e.g., the position of the first nucleotide in the Reference column |
|
Reference |
Sequence from the reference build, the first base starting at the base pair position in the Pos column |
|
Protein |
position |
Relative position of the amino acid in the protein |
Size |
Additional annotation using Ensembl lookup, based on protein (ENSP) |
|
VEP |
gene |
Ensembl stable ID of the affected gene |
impact |
||
Other columns |
Amino_acids |
Reference amino acid/substituted amino acid (in the case of a missense variant) |
Biotype |
Biological class of transcript or regulatory feature |
|
CallRatio |
Proportion of reads containing the variant call; expected to be close to 0.5 for heterozygous calls and close to 1 for homozygous calls |
|
cDNA_position |
Relative position of the base pair in the cDNA sequence |
|
CDS_position |
Relative position of the base pair in the coding sequence |
|
Codons |
The alternative codons with the variant base in uppercase |
|
Consequence |
Consequence type of this variant |
|
DISTANCE |
Shortest distance from variant to transcript (applies to up- and downstream variants) |
|
Depth |
The number of reads used in evaluating the corresponding call |
|
DOMAINS |
The source and identifer of any overlapping protein domains |
|
ENSP |
The Ensembl protein identifier of the affected transcript |
|
Existing_variation |
rs name of SNP if it exists |
|
EXON |
Ensembl (or Refgene) exon ID |
|
Feature |
Ensembl stable ID of feature |
|
Feature_type |
Type of feature, currently one of “Transcript”, “RegulatoryFeature”, or “MotifFeature” |
|
FILTER |
Quality parameter using the ratio between gt-quality and depth showing if the call is considered of “LowQual” quality (not useable) or “PASS”; this is still a very crude quality measure |
|
FS |
Fisher’s exact test of read strand; if the reference reads are balanced between forward and reverse strands, then the alternate reads should be as well |
|
formatZip |
VCF genotype field |
|
Gene_Symbol |
Based on HGNC when it exists, otherwise it is the Ensembl internal alias |
|
GL_Call |
A statistical measure indicating the likelihood that the call is wrong; the scale has been converted to using only integers - the higher the number, the less likely it is that the call is wrong |
|
GMAF |
MAF (minor allele frequency) of existing variant in Genomes 1000 Phase I |
|
HGNC |
The HGNC gene identifier |
|
HGVSc |
HGVSc coding sequence name |
|
HGVSp |
HGVSp protein sequence name |
|
INTRON |
The intron number (out of total number) |
|
Max_Consequence |
The consequence reported for this variant having the maximum impact |
|
Refgene |
Accession number from NCBI using lookup into Ensembl 69 using feature |
Perspective views¶
Perspectives subtabs focus on subsets of the columns in the Default view.
Perspective |
Description |
|---|---|
Basic |
|
Default view |
Shows all columns |