Related comments¶
The Related comments query retrieves comments in the local project based on a gene symbol, subject ID and, if desired, according to a user-defined threshold for the ACMG category 4 minimum allele frequency (Cat4minAF). The user may also designate a preferred VEP reference file (version). In addition, a relevance score (rscore) is calculated to indicate the degree of agreement between the phenotypes associated with the gene and those assigned to the subject (PN).
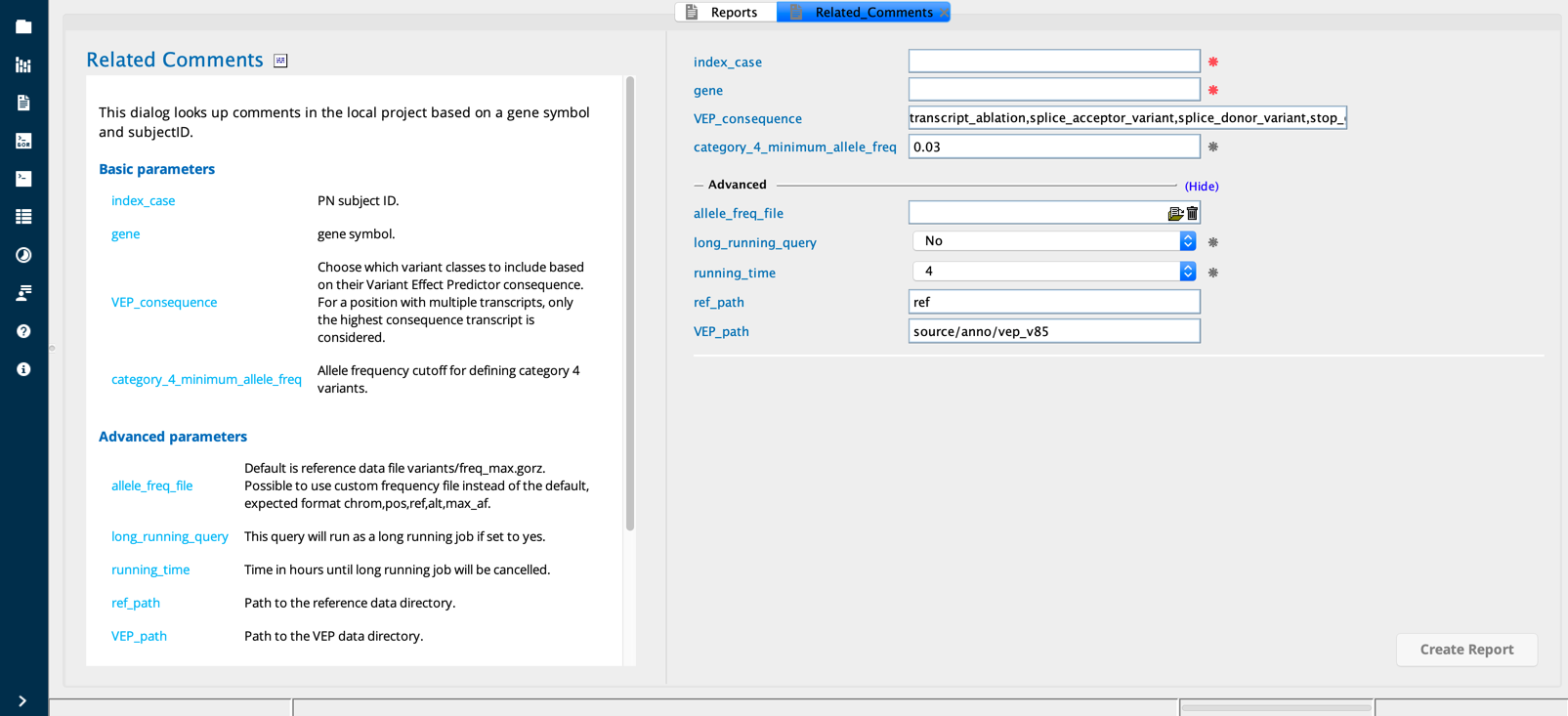
Related Comments module in Sequence Miner¶
Example use case¶
The user wishes to review and evaluate comments made by other investigators (in their institution’s project) on variants in a gene of interest.
Description of the algorithm¶
This query creates a table of all variants with comments in the project for the selected gene. For the indicated subject, the phenotype terms “present” and “absent” in the subject are reported as being present or absent in that individual.
A PN relevance score (rscore) is calculated as follows:
First the subject’s assigned HPO code(s) (phenotypes) are mapped onto a phenotype-ontology tree structure (parent-child relationship between terms) generated from the HPO database.
Next, the score is weighted by a generelevance score, a similar HPO code term score calculated based on the gene’s associated HPO terms.
Higher scores indicate greater agreement between the gene-variant-associated phenotype terms and the subject’s associated phenotype terms.
Interpreting the output¶
Each row lists a variant and its related comments, including text entered by the user, mode of inheritance, user ID, whether the variant is starred or not, etc. These columns correspond to the entries in the CSA variant annotation page, shown below.
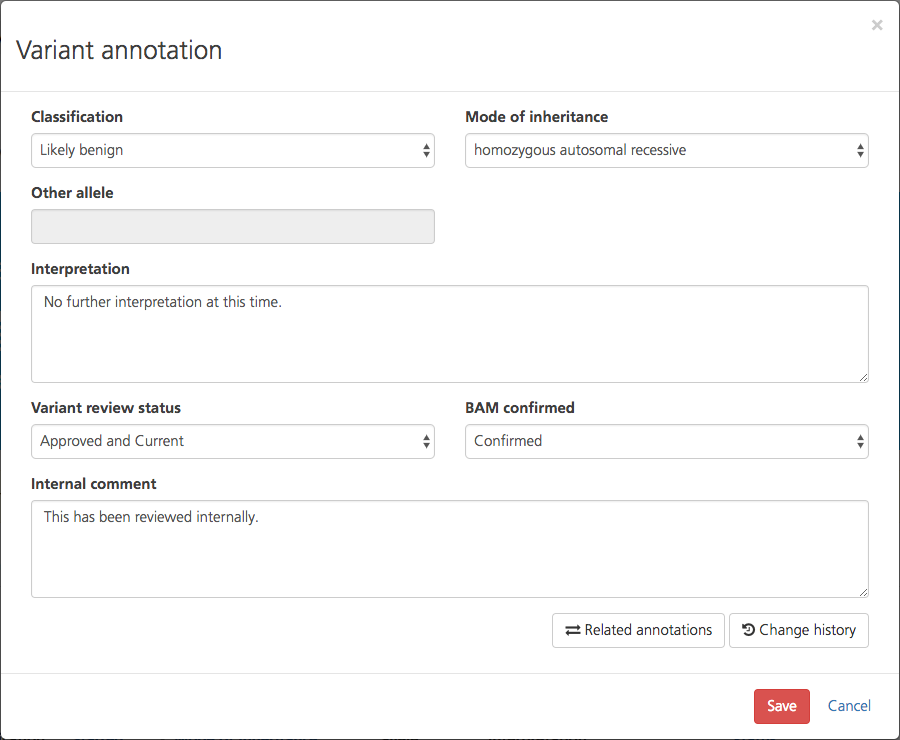
Variant view in CSA¶
Note
Columns displayed depend on the formats of the annotation files
Column descriptions¶
Group |
Column |
Description |
|---|---|---|
Basic |
alt |
|
chromo |
||
pn |
Subject ID |
|
pos |
||
ref |
||
gene |
prevalence_button |
|
symbol |
Based on HGNC when it exists, otherwise it is the Ensembl internal alias |
|
MAX |
AF |
Maximum allele frequency reported from the following databases: EVS, EXAC, THGP3, GONL, and KYOTO |
consequence |
||
Score |
||
other |
allele |
The alternative (reference) allele for the variant |
database_indicator |
||
pheno |
absent |
Phenotype terms for the gene that are not associated with the subject |
present |
Phenotype terms for the gene that are associated with the subject |
|
report |
section |
|
status |
||
type |
||
variant |
annotation_related |
|
review_status |
User note on whether the variant is in process or to be reviewed at a later date |
|
Other columns |
amcg_variant_annotation_id |
|
approval_months |
||
approved_clinical_significance |
||
bam_confirmed |
BAM file inspected to confirm the presence of the variant |
|
clinical_significance |
Observed clinical significance as reported in HGMD, OMIM, or ACMG |
|
computation_data_indicator |
||
DIAG_ACMGcat |
The ACMG category of the other variants (see DIAG_otherPos and DIAG_compPhases) that form compound heterozygosity with the variant |
|
disease |
The associated disease or phenotype |
|
extDiseaseDescription |
||
extDiseaseId |
||
hgvscp |
The HGVS coding/protein sequence name |
|
internal_comment |
Comment internal to the project |
|
interpretation |
||
is_active |
Yes (“1”) or no (“0”) |
|
last_approved_at |
||
mode_of_inheritance |
Observed mode of inheritance |
|
onset |
||
parental_origin |
||
population_data_indicator |
||
query_context |
Query filters used to identify variant |
|
reported_at |
||
reporting_category |
||
rscore |
Relevance score based on the degree of agreement between the phenotype terms signed to the subject and gene input for this query. The score is weighted by the distance of the gene and subject terms (parent/child branch distance) in the HPO ontology tree. High scores indicate greater agreement between the phenotypes associated with the subject and the gene variant. |
|
sanger_status |
Confirmed by Sanger sequence analysis or no |
|
severity |
||
study_id |
||
text |
Text added by a user in the comments section for each variant |
|
transcript |
||
transcript_raw |
||
transcriptFeature |
||
type |
||
user_id |
ID of the user |
|
VEP_Max_Impact |
Summary of Variant Effect Predictor consequence: HIGH is truncating; MODERATE is missense and splice site; LOW is synonymous, UTR, and intron; LOWEST is intergenic, promoter, and other |