Mendelian analysis¶
The Mendelian analysis dialog reports variants in a single index case that meet minimum filtering criteria and follow a Mendelian inheritance pattern in the family (if parents are available).
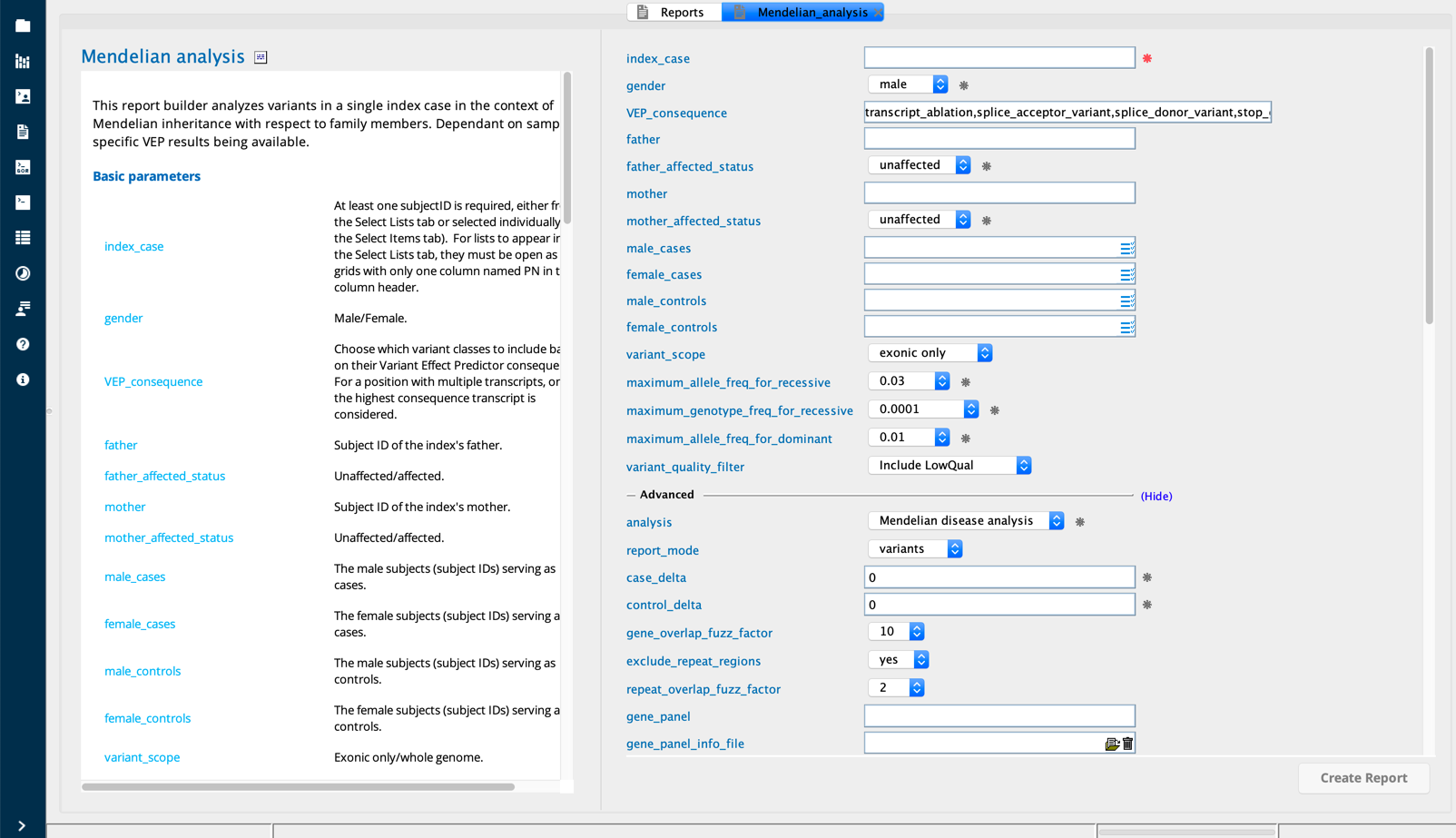
Mendelian analysis in Sequence Miner¶
Example use case¶
The user has a nuclear family that includes both unaffected parents and two siblings affected with a disorder. The user suspects a recessive genetic variant is the cause and wishes to screen family members for variants that are heterozygous in the parents and homozygous or compound-heterozygous in the affected siblings.
Description of the algorithm¶
For all selected family members, the query first extracts all variant calls that meet the filtering criteria (e.g., variants of user-defined known or predicted pathogenicity level, allele frequency or impact on the transcript, variants residing in user-selected genomic regions of inclusion or omission, etc.). Each variant is comprehensively annotated with allele frequency, reported or predicted level of pathogenicity, clinical information, and other attributes. The annotated variants are then presented in several different perspectives: AllByGene, AllVariants, and inheritance patterns in the subjects. The index case will be considered “affected” or a “case”. If the parents are unaffected, they will be treated as “controls”. Additional siblings added to the analysis will be considered “cases” if they are affected or “controls” if they are unaffected.
The inheritance model is designated as follows:
DIAG_HomRecess if the following criteria are met:
The index is homozygous for a variant meeting the user’s criteria OR if the variant is on the X chromosome, a male index is a hemizygous
Neither parent is homozygous OR if the variant is on the X chromosome and if the father is a carrier, then he is affected
DIAG_CHZ if the following criteria are met:
The index is a carrier of 2 different variants that meet the user’s criteria and reside in the same gene
The variants are each contributed by a different parent
DIAG_Dominant if the following criteria are met:
The index carries a variant with a max_Af < the user designated DmaxAf
The parent who carries the variant is affected
DIAG_denovo if the following criteria are met:
The index is a carrier of a variant meeting the user’s criteria and that has a VEP_max_Af < the user designated DmaxAf
Both parents have coverage depth > 8 reads at the locus
Both parents have fewer than 2 reads carrying the variant
Interpreting the output¶
The resulting output lists a single row per variant identified in the designated index case subject. The variants shown are filtered based on the criteria set by the user in the input parameters (e.g., call quality, read depth, allele frequency, VEP category, etc.). The variants are ordered by chromosome and position and annotated with coverage, quality metrics, variant counts by gene and affected status, as well as information from public clinical databases.
Based on the expected pattern of inheritance, the user may begin by reviewing the results in the corresponding inheritance perspective (e.g., ARhomAndCHZ - autosomal recessive and compound heterozygous variants). Alternatively, the review may start in the AllVariants perspective where the user can filter various columns by right-clicking the column header to select the filtering threshold for the selected column/attribute (e.g., columns for CASESwithHomoVar, FATHERwithVar, MOTHERwithVar).
Column descriptions¶
Group |
Column |
Description |
|---|---|---|
Basic |
||
Call |
||
CHROM |
||
hetORhom |
||
PN |
||
POS |
||
Reference |
Group |
Column |
Description |
|---|---|---|
CGD |
The CGD columns provide information for variants based on the manually curated database of variants associated with known medically significant conditions and available interventions |
|
AGE_GROUP |
Pediatric: less than 18 years of age; Adult: At least 18 years of age |
|
COMMENTS |
Any additional observations noted by curators |
|
CONDITION |
Conditions resulting from mutations in the same gene but may otherwise be placed in the “General” Intervention category |
|
INHERITANCE |
Pattern of inheritance: AD - autosomal dominant; AR - autosomal recessive; BG - blood group; Digenic - A condition resulting from simultaneous mutations in two different genes; Maternal - maternal mitochondrial inheritance; XL - X-linked (because X-linked conditions can frequently have manifestations in both genetic sexes, X-linked conditions are not designated as dominant or recessive) |
|
INTERVENTION_CATEGORIES |
Includes organ systems for which specific and additional inteventions may be beneficial |
|
INTERVENTION RATIONALE |
Description of the intervention and its benefit |
|
MANIFESTATION CATEGORIES |
Includes organ systems affected by mutations in corresponding genes; recognition of involved organ systems may help guide supportive care |
|
REFERENCES |
The pubmed ID (PMID) of the referenece(s) |
Group |
Column |
Description |
|---|---|---|
COMM |
COMM annotation is comprised of comments added by users in the project and comments drawn from the |
|
amcg_variant_annotation_id |
||
approval_months |
Counter for number of months since the date of last approved clinical significance |
|
approved_clinical_significance |
The clinical significance approved by the user |
|
bam_confirmed |
The user’s notation if the BAM files were reviewed to confirm the presence/absence of the variant |
|
clinical_significance |
The clinical significance (e.g., pathogenic, benign, unknown significance, drug-response, risk factor, etc.) of the variant as annotated (commented) by users; if the same variant has several comments, this cell will contain a set of values |
|
computation_data_indicator |
||
created_at |
Date and time stamp the comment was created |
|
disease |
The associated disease or phenotype |
|
extDiseaseDescription |
||
extDiseaseId |
||
extDiseaseName |
||
gene_prevalence_button |
||
gene_symbol |
Gene symbol from the |
|
hgvscp |
The HGVS coding/protein sequence name |
|
id |
Comment ID |
|
internal_comment |
User comment created for internal use |
|
interpretation |
Interpretation on a variant, added by the user |
|
is_active |
User comment on Active status of the variant |
|
last_approved_at |
The last time the variant was approved by the user |
|
mode_of_inheritance |
The user-annotated (commented) mode of inheritance of the variant; if the same variant has several comments, this cell will contain a set of values |
|
onset |
The onset of the disease the variant is associated with, curated by the user |
|
other_allele |
User comment on the other allele |
|
other_databases_indicator |
||
owner |
User name |
|
parental_origin |
Parental origin of the variant selected by the user, e.g., father, mother, both, de novo, unknown, etc. |
|
pn |
User comment on the patient |
|
population_data_indicator |
||
query_context |
||
report_section |
Section in which the variant should be placed, selected by the user |
|
report_status |
Indicates what to report, related to client workflow |
|
report_type |
Type of report, related to client workflow |
|
reported_at |
Date of variant annotation |
|
reporting_category |
Reporting category selected by the user, related to client workflow |
|
sanger_status |
User comment on the status of the confirmation by Sanger sequencing |
|
STARRED |
A Boolean column indicating if the variant has been marked (starred) by users |
|
severity |
Severity of the disease with which the variant is associated, added by the user |
|
text |
The description (comment) component for the user annotation of the variant |
|
transcript |
||
transcript_raw |
All possible transcripts with HGVS nomenclature for the variant that can be selected by the user |
|
transcriptFeature |
||
type |
||
user_id |
User ID |
|
variant_annotation_related |
||
variant_review_status |
Variant review status added by the user |
Group |
Columns |
Description |
|---|---|---|
DIAG |
The DIAG columns provide diagnostic information for categorizing variants in a clinical setting based on the American College of Medical Genetics and Genomics (ACMG) guidelines. For reference, “diagnosis” is abbreviated as “dx” in some cases. |
|
ACMGcat |
Categorization of the sequence variants according to the ACMG scheme |
|
CHZ |
This field is equal to 1 (true) if the index is/cases are compound heterozygous (chz) (see GENE_CHZinGene) and none of the controls are homozygous |
|
deNovo |
A Boolean column indicating if a variant is de novo, i.e., not present in the parents |
|
Dominant |
The dominant variant; field is “true” if the variant is not present in unaffected individuals but is present in affected individuals |
|
HomRecess |
The homozygous recessive variant; field is “true” if the variant is heterozygous in unaffected individuals but is present in affected individuals |
|
model |
Autosomal or X-linked analysis |
|
otherACMGcat |
The ACMG category of other variants (see DIAG_otherPos and DIAG_compPhases) that form compound heterozygosity with the variant |
|
otherPos |
For a particular CHZ variant, the base pair position of the other variant(s) that produces compound heterozygosity in a gene |
|
rank_dominant |
An emperical ranking of variants. Low-ranking variants are likely to be better candidates for dominant variants than those with a high rank. This ranking takes into consideration how many affected individuals share the same variant or any variants in the same gene, and how few controls have the variant or any variant in the same gene (see also DIAG_Dominant). |
|
rank_recessive |
An emperical ranking of variants. Low-ranking variants are likely to be better candidates for recessive variants than those with a high rank. The ranking takes into consideration homozygosity and compound heterozygosity in the affected vs. unaffected individuals. Furthermore, the smaller the DIAG_recessiveCat value (e.g., “1:DxConsistent”, “2:DxLikely”, “3:DxPossible”, etc.), the larger the weight of the variant. |
|
recessiveCat |
|
Group |
Columns |
Descriptions |
|---|---|---|
EuroGenetest |
The EuroGenetest columns are derived from a European Commission project database containing European genetic testing information for particular genes, variants, and diseases. |
|
Diseases |
Diseases associated with a variant derived from the European Commission project database |
|
NoOfDiseases |
Number of diseases associated with a variant derived from the European Commission project database |
|
NoOfpanels |
Number of gene panels associated with a variant derived from the European Commission project database |
|
panels |
EuroGenetest panels associated with a variant derived from the European Commission project database |
Group |
Columns |
Description |
|---|---|---|
FATHER |
The FATHER columns provide variant annotations for the father. The same attributes are listed for the mother in the MOTHER group of columns. |
|
apprCovDepth |
The approximate read depth of the given variant |
|
call |
Sequence (variant) called based on the reference sequence at the designated position |
|
hetORhom |
Heterozygous or homozygous of given variant |
|
homozVarsInGene |
Equal to 1 if the father has some homozygous variant in the given gene |
|
knownVarsInGene |
The number of known variants (e.g., in HGMD, ClinVar, OMIM) that the father has in the given gene |
|
readsWithVar |
The number of reads contains the variant allele |
|
subjCompHeterInGene |
Equal to 1 if the father has potentially compound heterozygous for a given gene, otherwise it is equal to 0. Compound heterozygosity requires the case to have a single homozygous variant or two heterozygous variants in a given gene. |
|
subjWithVarInGene |
Equal to 1 if the father has variant in the given gene, otherwise it is equal to 0 |
|
withHomoVar |
Equal to 1 if the father is homozygous for a given variant |
|
withVar |
Equal to 1 if the father has the variant, otherwise it is equal to 0 |
Group |
Columns |
Description |
|---|---|---|
GENE |
The GENE columns provide information based on the candidate gene in which a variant is found. When possible, the HUGO Gene Nomenclature Committee (HGNC) gene symbol is provided. |
|
Aliases |
Aliases of a given gene as annotated by Ensembl |
|
avg_depth |
The average sequence read depth in the exome of a given gene |
|
candidate_paralogs |
If the gene is a paralog of a designated candidate gene, then it is listed in this column |
|
CandorParalog |
“c” indicates the gene is a designated candidate gene and “p” indicates the gene is a paralog of a designated candidate gene |
|
cov |
The fraction of genes (exome) with depths <5 (L), ranging from 5-9 (M), and greater than 9 (H); the scale is from 0 to 1 |
|
DmaxAf |
The maximum allowed allele frequency for a dominant variant |
|
exomeSize |
The sum of exon in the given gene |
|
exontype |
The type of exon (“coding” or “noncoding”) |
|
homozVarsInGene |
The number of homozygous variants the index case has in a given gene |
|
iheVarsInGene |
The number of inheritance error variants the index case has in a given gene |
|
knownVarsInGene |
The number of known (e.g., in HGMD) variants the the index case has in a given gene |
|
lt10 |
The fraction of the exome with sequence read coverage less than 10X |
|
lt15 |
The fraction of the exome with sequence read coverage less than 15X |
|
lt20 |
The fraction of the exome with sequence read coverage less than 20X |
|
lt25 |
The fraction of the exome with sequence read coverage less than 25X |
|
lt30 |
The fraction of the exome with sequence read coverage less than 30X |
|
lt5 |
The fraction of the exome with sequence read coverage less than 5X |
|
maximum_allele_freq_for_dominant |
||
maximum_allele_freq_for_recessive |
||
maximum_genotype_freq_for_recessive |
||
MOI |
Mode of inheritance |
|
Paralogs |
The paralogs of a given gene |
|
symbol |
Gene identified by viewing variant in Ensembl; outputs HGNC gene symbol for gene identified (clone name provided when HGNC unavailable) |
|
varsInGene |
The total number of variants in a gene for the index case |
Group |
Column |
Description |
|---|---|---|
GO |
The GO columns provide a functional annotation of the gene product in which the variant is found |
|
Descriptions |
Gene ontology descriptions |
|
IDs |
Gene ontology identifiers |
Group |
Column |
Description |
|---|---|---|
GT |
The GT (genotype attributes) gruop of columns provide quality control information for the variant call based on the sequence read depth and quality |
|
CallCopies |
Refers to how many copies of the variation exist in a subject. A CallCopies value of “2” therefore corresponds to a homozygous variant; a CallCopies value of “1” corresponds to a heterozygous variation; a CallCopies of “0” corresponds to No Call |
|
CallRatio |
Proportion of reads containing the variant call; expected to be approximately 0.5 for heterozygous calls and close to 1 for homozygous calls |
|
Depth |
The number of reads used for evaluating the corresponding call |
|
FILTER |
Quality parameter using the ratio between gt-quality and depth, showing if the call is considered “LowQual” quality or “PASS” |
|
GL_Call |
A statistical measure indicating the likelihood that the call is wrong; the scale has been converted to use only integers - the higher the number, the less likely it is that the call is wrong |
|
IHEstatus |
A Boolean column (1/0) indicating whether the variant is an inheritence error or not; “1” indicates that the variant is an inheritance error, “0” indicates that the variant is not an inheritance error |
Group |
Column |
Description |
|---|---|---|
KNOWN |
The KNOWN columns provide publicly available information about the candidate gene and/or variant as annotated by HGMD, ClinVar, and OMIM. |
|
ClinVarAcc |
The associated ClinVar accession ID |
|
distance |
The distance between a known pathogenic variant (Cat1, pathogenic annotation in HGMD, ClinVar, or OMIM) and the identified variant |
|
exactMatch |
A Boolean column (1/0) indicating if the variant (chromosome, position, reference, call) is a direct match to a known pathogenic variant (instead of near a known variant, or at the same position with a different call allele) |
|
Gene_diseases |
Diseases known to be associated with the gene as annotated by HGMD, ClinVar, and OMIM |
|
Gene_par_diseases |
Diseases known to be associated with the parlogs of the gene as annotated by HGMD, ClinVar, and OMIM |
|
GeneLists |
Pre-defined gene list membership of the gene in which the variant is found |
|
HGMDacc |
The related HGMD accession ID |
|
InACMG |
A Boolean column (1/0) indicating whether the gene is in the ACMG recommended list for incidental findings |
|
pmid |
Pubmed ID of the reference from which the information was obtained |
|
source |
The public database (ClinVar, HGMD, or OMIM) source for annotation |
|
var_diseases |
Diseases known to be associated with the variant as annotated by HGMD, ClinVar, and OMIM |
|
variantType |
The type of the variant (germline, pathogenic, etc.) |
Group |
Columns |
Descriptions |
|---|---|---|
MOTHER |
The MOTHER columns provide variant annotations for the mother. The same attributes are listed for the father in the FATHER group of columns. |
|
apprCovDepth |
The approximate read depth of the given variant |
|
call |
Sequence (variant) called based on the reference sequence at the designated position |
|
hetORhom |
Heterozygous or homozygous of given variant |
|
homozVarsInGene |
Equal to 1 if the mother has some homozygous variant in the given gene |
|
knownVarsInGene |
The number of known variants (e.g., in HGMD, ClinVar, OMIM) that the mother has in the given gene |
|
readsWithVar |
The number of reads contains the variant allele |
|
subjCompHeterInGene |
Equal to 1 if the mother has potentially compound heterozygous for a given gene, otherwise it is equal to 0. Compound heterozygosity requires the case to have a single homozygous variant or two heterozygous variants in a given gene. |
|
subjWithVarInGene |
Equal to 1 if the mother has variant in the given gene, otherwise it is equal to 0 |
|
withHomoVar |
Equal to 1 if the mother is homozygous for a given variant |
|
withVar |
Equal to 1 if the mother has the variant, otherwise it is equal to 0 |
Group |
Column |
Description |
|---|---|---|
OMIM |
The OMIM columns provide the OMIM-designated identification for a particular gene and related disease description. |
|
Descriptions |
OMIM disease descriptions for the gene |
|
IDs |
The OMIM ID of the gene |
Group |
Column |
Description |
|---|---|---|
VEP |
The VEP (Variant Effect Predictor) columns provide functional annotations for variants based on the ENSEMBL SNP Effect Predictor database. For more information, visit the VEP web page: http://www.ensembl.org/info/docs/tools/vep/index.html/. |
|
Amino_acids |
The amino acid with and without variant (only provided if the variation affects the protein-coding sequence), otherwise “.” |
|
Biotype |
Biological class of transcript or regulatory feature |
|
CDS_position |
Position of the base pair in the coding sequence; a value is given for each transcript |
|
Consequence |
The Variant Effect Predictor annotation of the consequence type in the canonical transcript of the annotated variant |
|
Max_Af |
Maximum reported allele frequency across the population surveys from 1000GP3, EVS, EXAC, Kyoto, GONL (Variant View - Frequencies) |
|
Max_Consequence |
VEP predicted consequence for a variant producing the the greatest impact on the transcript |
|
Max_Impact |
Classification of the level of severity of the transcript consequence type assigned by VEP |
|
Max_Score |
Maximum score for the variant as observed in dbNSFP [Score=max ((1-Sift_score), Polyphen2_HDIV_score, Polyphen2_HVAR_score)]. |
|
Protein_position |
Position of the amino acid in the protein sequence (only if the variant falls within a coding sequence); a value is given for each corresponding transcript specified in the CDS position field |
|
Refgene |
The accession number from NCBI of the affected transcripts |
|
Transcript_count |
The number of different transcripts in which the variant is found |
Group |
Column |
Description |
|---|---|---|
Other columns |
||
Biotype |
Additional annotation using Ensembl lookup based on feature, e.g., miRNA_PUTATIVE, misc_RNA_PUTATIVE, protein_coding_PUTATIVE, pseudogene_PUTATIVE, snoRNA_PUTATIVE |
|
Carrier |
Indicates if the individual carries the variant (“true” or “false”) |
|
CLNACC |
ClinVar assigned variant accession number |
|
formatZip |
VCF genotype fields |
|
FS |
Fisher’s exact test of read strand; if the reference reads are balanced between forward and reverse strands, then the alternate reads should be as well |
|
HGVSc |
The HGVS coding sequence name |
|
HGVSp |
The HGVS protein sequence name |
|
ID |
Identifier, either hgmd or pmid/omim in clinical detail map |
|
Refgene |
mRNA accession number from NCBI using lookup into Ensembl using feature |
|
rsIDs |
rsID of the SNP entry in dbSNP, corresponding to the mutation, where available |
|
Sequence_Variant |
The formatted sequence variant |
|
Source |
This column lists the parent/source (one parent) of the corresponding row variant |
|
Sourcex |
This column lists the other parent/source of the corresponding row variant |
|
varType |
“del” for deletion, “ins” for insertion, and “sub” for substitution |
Perspective views¶
The Default view perspective shows all of the columns described above. Additional perspectives focus on subsets of the columns in the default view.
Perspective |
Description |
|---|---|
ACMG |
|
AllByGene |
Focuses on all variants that meet the user’s criteria and all genes carrying such variants |
AllKnownVariants |
|
AllVariants |
Focuses on all variants that meet the user’s criteria and all genes carrying such variants |
ARhomAndCHZ |
Focuses on all variants that meet the user’s criteria and that fit the corresponding inheritance models |
AutoRecessHom |
Focuses on all variants that meet the user’s criteria and that fit the corresponding inheritance models |
CandidateGenesByGene |
Focuses on variants that meet the user’s criteria and are in the user’s selected candidate genes (when applicable) |
CandidateGenesByVar |
Focuses on variants that meet the user’s criteria and are in the user’s selected candidate genes (when applicable) |
CommentedVariants |
All variants in the index that meet the user’s criteria and that have been previously annotated by a user |
CompoundHetero |
Focuses on all variants that meet the user’s criteria and that fit the corresponding inheritance models |
Default view |
Shows all columns |
DeNovoInCandidates |
Focuses on variants that meet the user’s criteria and are in the user’s selected candidate genes (when applicable) |
Dominant |
Focuses on all variants that meet the user’s criteria and that fit the corresponding inheritance models |
gene_panelGenesByGene |
Focuses on variants that meet the user’s criteria and are in genes from the user’s selected gene panel (or paralogs). When the user enters a gene_panel as an optional parameter, results will be displayed only for variants in those genes |
gene_panelGenesByVar |
Focuses on variants that meet the user’s criteria and are in genes from the user’s selected gene panel (or paralogs). When the user enters a gene_panel as an optional parameter, results will be displayed only for variants in those genes |
KnownVariants |
focuses on variants that meet the user’s criteria and are known pathogenic variants reported in HGMD, OMIM, or ClinVar |
MissKnownDiseaseGenes |
|
MissKnownVarsInCandGenes |
|
OMIM |
|
PossMissInCandGenes |
|
XlinkedRecess |
Focus on all variants that meet the user’s criteria and that fit the corresponding inheritance models |
Drill-in reports¶
Drill-in |
Description |
|---|---|
CaseAndControl_Variants |
For the selected gene variant in the Mendel report, this drill-in report lists the variant genotype for each carrier |
CaseAndControl_VarsInGene |
For the selected gene variant in the Mendel report, this drill-in report lists all case and controls variants in the same gene with any of the user-selected maximum consequences and the variant genotype for each carrier |
CaseAndControl_AllVarsInGene |
For the selected gene variant in the Mendel report, this drill-in report lists all variants identified in the same gene as the selected gene variant and the genotype for each carrier |