Loss of heterozygosity¶
The Loss of heterozygosity dialog identifies genomic or exonic regions exhibiting loss of heterozygosity in selected subjects, based on a user-defined region size and minimum variant count.
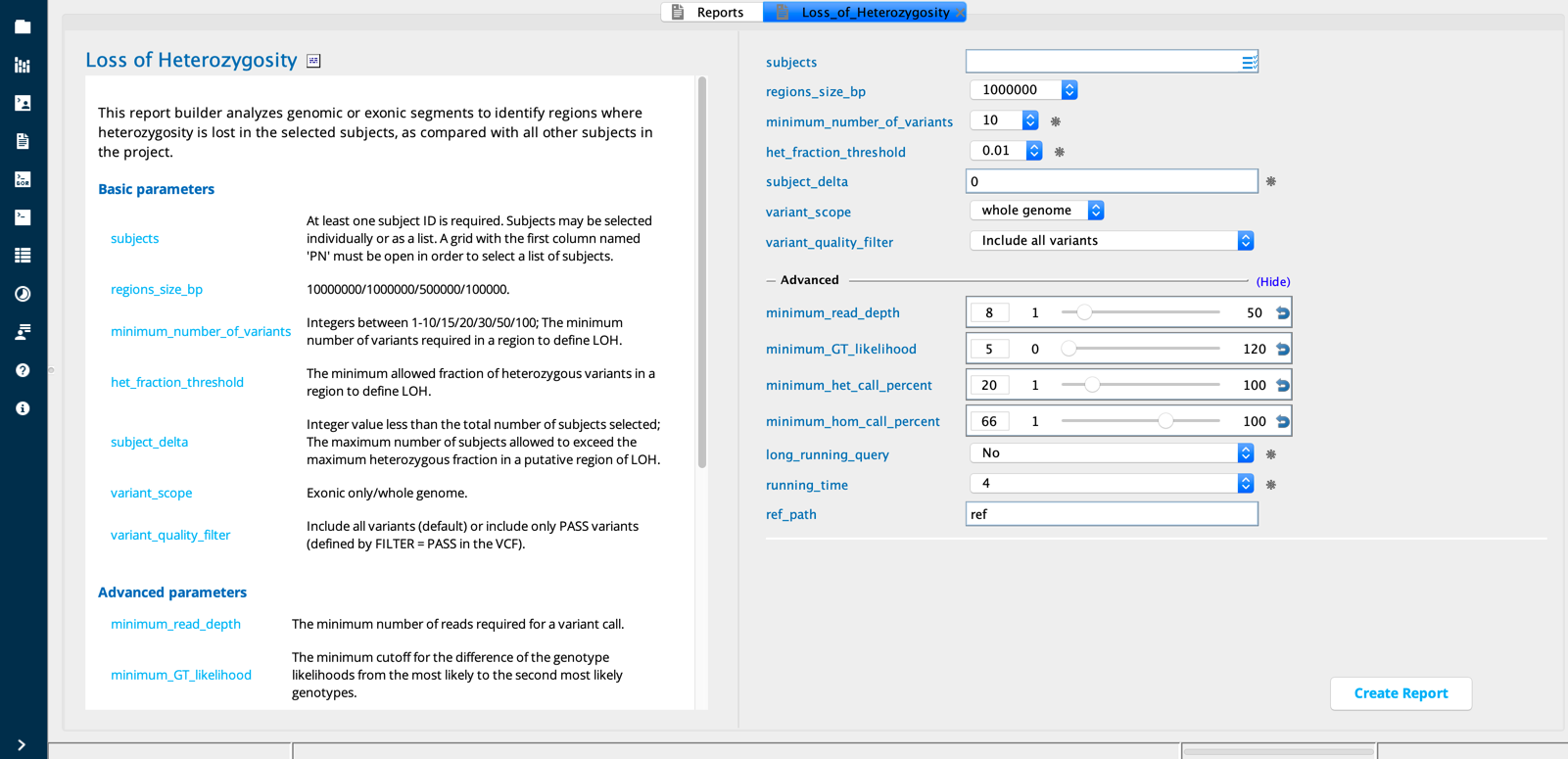
Loss of Heterozygosity module in Sequence Miner¶
Example use case¶
The user suspects Lynch syndrome in a multiplex family with colorectal cancer and wishes to screen four Lynch-associated genes for LOHZ in these family members. This report builder measures loss of heterozygosity across the whole genome or coding sequence based on the user-determined LOHZ-region size threshold, penetrance, and read quality parameters.
Description of the algorithm¶
The LOHZ algorithm measures heterozygosity across the genome in blocks of the selected size threshold. For the selected subjects, the number of variants (heterozygous and homozygous) within the defined region size are summed. The heterozygous calls are separately summed across each region block.
Based on the maximum heterozygous fraction (0, 1%, 2%, 5%, or 10%) defined by the user, the total number of variants is multiplied by this fraction and compared to the sum of the heterozygous variants in this region. If the sum of heterozygous variants is less than the fraction of total variants, then the region is marked as having loss of heterozygosity (LOHZ).
Finally, the regions with LOHZ are summed across the patients. If the LOHZ region is shared by all selected patients (or fewer depending on the value entered in the subject_delta field) and the total number of variants in the region exceeds the minimum number of variants defined by the user, then the region is reported as having LOHZ. The region, region size, number of variants in the region, number of patients that share the LOHZ region, and the genes found in this region are reported in the output.
Interpreting the output¶
The final report contains a single row for each region identified as having LOHZ. The genomic coordinates (Chrom, bpStart, and bpStop) are in columns 1-3 of each row, and are used to calculate the LOH region size (LOHregionSize column). The number of variants used in the calculation and the subjects exhibiting LOHZ are listed in the VariantsinRegion and numLOHZsubjects columns respectively. The final column provides a comma-separated list of the genes found within the region of LOHZ.
Column descriptions¶
Column |
Description |
|---|---|
bpStart |
The start position of the region |
bpStop |
The stop position of the region |
Chrom |
The chromosome of the region |
Gene_Symbol |
Based on HGNC when it exists, otherwise it is the Ensembl internal alias |
LOHregionSize |
The size of the LOHZ region calculated from bpStop - bpStart |
numLOHZsubjects |
The number of subjects/individuals/samples that have the region with LOHZ |
VariantsInRegion |
The number of variants (heterozygous and homozygous) in the region |