Internal annotations¶
The Internal annotations report builder compiles and annotates a list of variants or genes with variants in the project that have been commented on in Clinical Sequence Analyzer (CSA) or Sequence Miner (SM). Annotation is based on the selected reportMode (byGene or byVariant) and whether the user selects “yes” or “no” to includePhenotypes.
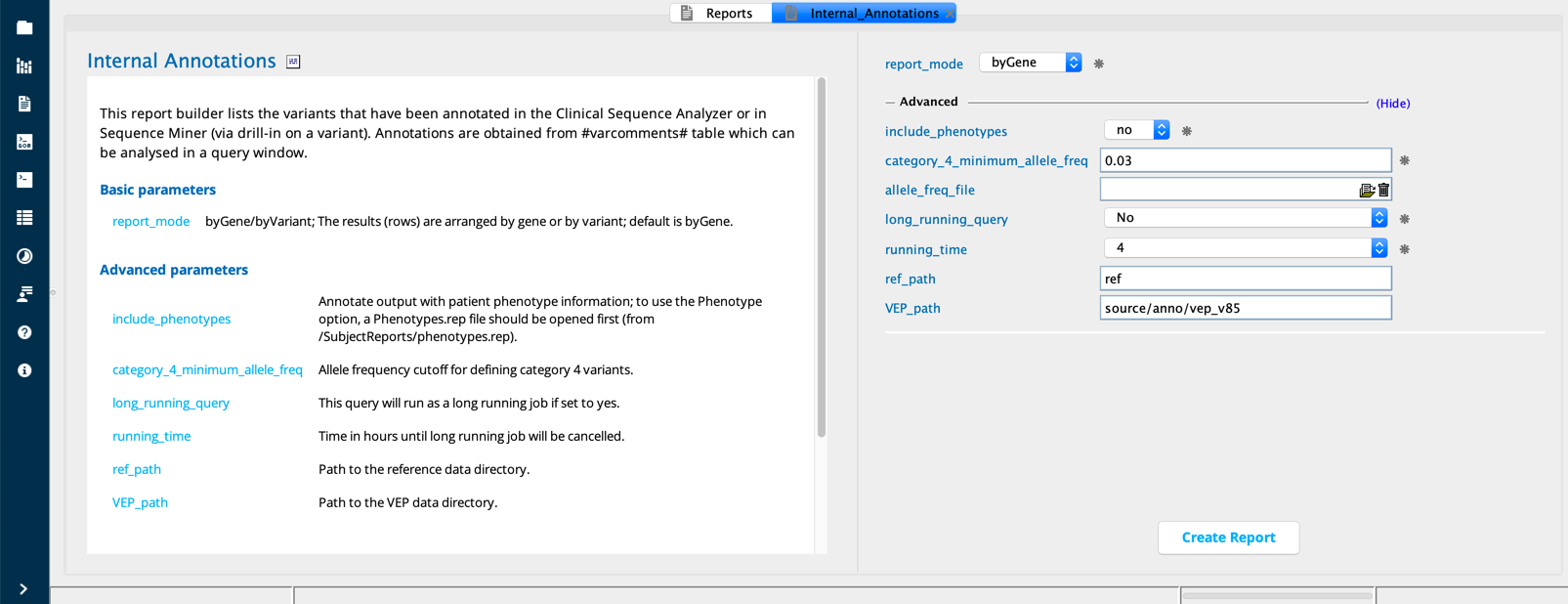
Internal Annotations module in Sequence Miner¶
Example use case¶
The user wishes to compile a list of all variants in the project that are related to cardiomyopathy and that have been annotated (commented on) by all users in the project. The user runs the Comments report builder setting reportMode to “byVariant” and selects “yes” to includePhenotypes. The user then filters the description column on cardiomyopathy.
Description of the algorithm¶
Variants or genes with variants that have been commented on internally (by users in the project) are retrieved and joined with corresponding annotation tables based on the selected report format.
Interpreting the output¶
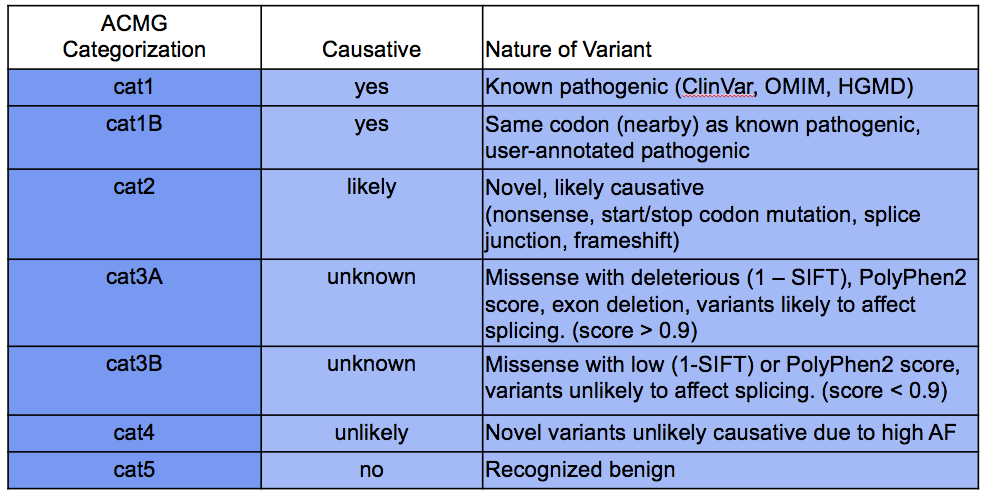
Column headers for annotations are described in the tables below. Annotation from numerous sources are provided. These include VEP, allele frequency and ACMG Category for variants and GO, OMIM, ClinVar and HGMD for genes.
Column descriptions¶
Column |
Description |
|---|---|
Chrom |
The chromosome of the variant represented as chr1, chr2, …, chr22, chrXY, chrX, chrY, chrM |
gene_symbol |
Based on HGNC when it exists, otherwise it is the Ensembl internal alias |
set_clinical_significance |
Clinical significance (e.g., pathogenic, benign, unknown significance, drug-response, risk factor, etc.) of the variant as annotated (commented) by users; if the same variant has multiple associated comments, this cell will contain a set of values |
set_mode_of_inheritance |
User annotated (commented) mode of inheritance of the variant; if the same variant has multiple associated comments, this cell will contain a set of values |
code |
HPO or OMIM taxonomy code for designated phenotype (shown only when includePhenotypes is set to “yes”) |
description |
HPO or OMIM description for designated phenotype (shown only when includePhenotypes is set to “yes”) |
Column |
Description |
|---|---|
gene_start |
The start base-pair position of the gene (zero based, i.e., the position of the base-pair before the first base-pair in the gene) |
gene_end |
Coordinate of the last nucleotide on the gene |
GENE_Aliases |
The aliases of the given gene |
GENE_Paralogs |
The paralogs of the given gene |
GO_IDs |
Gene Ontology identifiers |
GO_Descriptions |
Gene Ontology category descriptions |
KNOWN_Gene_diseases |
Diseases known to be associated with the gene as annotated by HGMD, ClinVar, and OMIM |
KNOWN_lis_disease |
Diseases known to be associated with the gene as annotated by HGMD, ClinVar, and OMIM |
KNOWN_MaxClinImpact |
The maximum clinical consequence of the variant (pathogenic, unknown etc) annotated by any one of HGMD, OMIM or ClinVar |
OMIM_IDs |
OMIM ID of the gene |
OMIM_Descriptions |
OMIM disease descriptions for the gene |
dis_PN |
Number of carriers of this variant with comments |
Column |
Description |
|---|---|
pos |
The (first) basepair position of the sequence variant, e.g., the position of the first nucleotide in the Reference column |
ref |
Reference allele in vcf format |
alt |
Alternative allele in vcf format |
VEP_Amino_Acids |
The amino acid with and without variant (only provided if the variant affects the protein-coding sequence), otherwise “.” |
VEP_Biotype |
Biological class of transcript or regulatory feature |
VEP_CDS_position |
Position of the base pair in the coding sequence; a value is given for each transcript |
VEP_max_consequence |
VEP predicted consequence for a variant producing the the greatest impact on the transcript |
VEP_Max_Impact |
Classification of the level of severity of the transcript consequence type assigned by VEP |
VEP_Max_Score |
Maximum score for the variant as observed in dbNSFP [Score=max ((1-Sift_score), Polyphen2_HDIV_score, Polyphen2_HVAR_score)] |
VEP_Protein_Position |
Position of the amino acid in the protein sequence (only if the variant falls within a coding sequence); a value is given for each corresponding transcript specified in the CDS position field |
VEP_Refgene |
The Accession number from NCBI of the affected transcripts |
VEP_Transcript_count |
Number of different transcripts in which the variant is found |
MAX_AF |
Maximum reported allele frequency (1000GP3, EVS, EXAC, Kyoto, GONL) |
set_pn |
IDs of a variant carriers |
DIAG_ACMGcat |
Categorization of the likelihood pathogenicity of sequence variants according to the ACMG scheme; values range from cat1 (most severe) to cat4 (least severe). See table below for ACMG categorization descriptions. |