Exon coverage¶
Exon Coverage module in Sequence Miner¶
Example use case¶
The Exon coverage query reports sequence read coverage and additional attributes for each exon. One or more subjects can be selected, and results are reported separately for each subject. The analysis is based on a pileup from the original BAM files and is presented in terms of the following:
The average depth of coverage over each exon, on a per subject basis
The fraction of each exon with coverage below specific thresholds; specifically the fraction of each exon with coverage less than 5, less than 10, and so on up to 30
Exonic regions are defined by Ensembl or RefGenes (both are obtained from the VEP and Ensembl release shown in the version.txt file in the ref directory, or the Version Information link in CSA). Selection between the Ensembl and RefGene reference gene sets is determined by the gene reference set input argument.
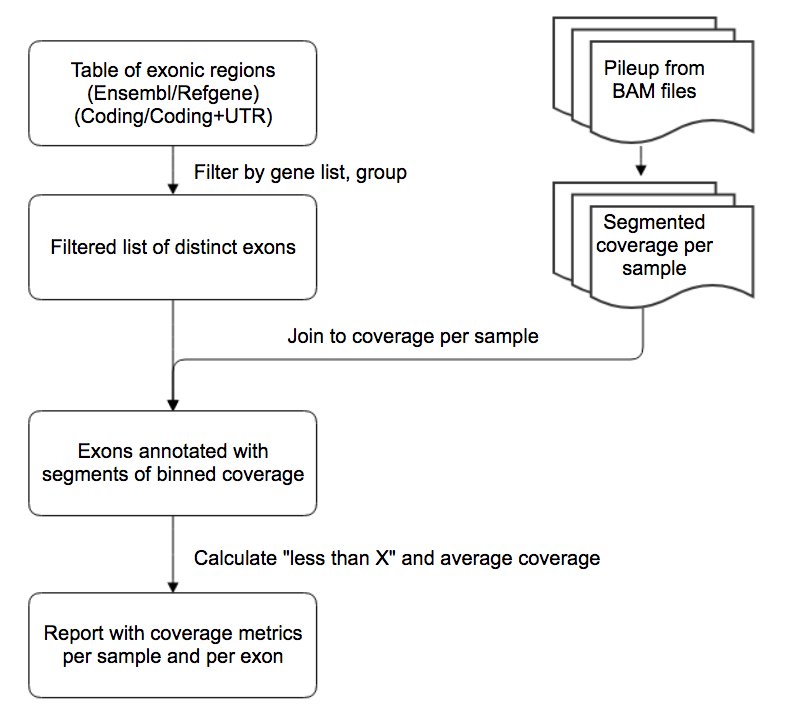
Approximating the coverage per exon based on segment coverage¶
When BAM files for a sample are imported into the WuXi NextCODE system, a segment coverage file ( segment_cov.gord ) is generated with the total read depth for each base. This depth is binned to the nearest interval to reduce file size and improve query speed. Therefore, the results from this report builder are slightly approximated.
Limiting the input list of subjects to maintain a reasonable output report size¶
The output report can be quite large, with a total number of rows proportionate to the number of input PNs. In order to avoid “out of memory” issues on the local computer, the number of input subjects is limited to 20 or fewer. In order to analyze a larger number of subjects, the report can be run multiple times on batches of 20.
Description of the algorithm¶
Interpreting the output¶
Each row of output lists attributes and quality statistics per exon for each gene of interest in a single subject (PN). These values include the genomic coordinates of the exon, the exome size (length of the exon in bp), average depth over the exon, Ensembl gene and transcript annotation, the biotype of the exon, and a breakdown of the proportion of the exon with different degrees of coverage. The metrics used to flag regions of low coverage are lt5, lt10, lt15, lt20, lt25, and lt30. Each metric indicates a given coverage threshold, with values between between 0 and 1. For instance, a value of “0.05” for lt10 indicates that 5% of the exon is covered by fewer than 10 sequence reads, and therefore 95% of the exon has at least 10X coverage.
Input parameters¶
Input |
Description |
Values |
|---|---|---|
genome_range |
All (whole genome) / selected region (from an open Genome Browswer window) |
Inputs are defined by any open Genome Browser window |
subjects |
Subjects may be selected individually or as a list |
A grid with the first column named “PN” must be open in order to select a list of subjects |
gene_list |
Filter to include only variants in the gene list |
The list should be open in a grid tab containing a single column of gene symbols with a header labeled “gene_sybmol” |
exon_reference |
Choose whether to limit to coding exons |
Select Coding only or Coding and UTR |
gene_reference_set |
Choose between Ensembl or RefGene reference sets |
Select Ensembl or RefGene |
Input |
Description |
Values |
|---|---|---|
long_running_query |
This query will run as a long running job if set to “Yes” |
Yes or No |
running_time |
Time in hours until long running job will be cancelled |
1, 2, 4, 8 hours |
ref_path |
Path to the reference data directory |
Default: ref |
Column descriptions¶
Group |
Column name |
Description |
|---|---|---|
Basic |
Chrom |
Chromosome |
pn |
The subject ID (identifier) |
|
Gene |
biotype |
Biological class of gene as annotated by Ensembl |
stable_id |
Ensembl stable ID for the gene |
|
symbol |
Based on HGNC when it exists, otherwise it is the Ensembl internal alias |
|
Other columns |
avg_depth |
The average read depth of the specific exon |
Exon_size |
The number of base pairs comprising the given exon |
|
exon |
Ensembl exon ID |
|
exonend |
Coordinate for the last base of the exon |
|
exonstart |
Coordinate for the first base of the exon |
|
lt10 |
The fraction of the exome with sequence read coverage less than 10X |
|
lt15 |
The fraction of the exome with sequence read coverage less than 15X |
|
lt20 |
The fraction of the exome with sequence read coverage less than 20X |
|
lt25 |
The fraction of the exome with sequence read coverage less than 25X |
|
lt30 |
The fraction of the exome with sequence read coverage less than 30X |
|
lt5 |
The fraction of the exome with sequence read coverage less than 5X |
|
set_Transcript_stable_id |
Comma-separated list of Ensembl transcript IDs related to the specific exon |
|
strand |
Plus/minus of the DNA strand encoding the exon sequence |
|
Transcript_biotype |
Biological class of transcript |