De novo/CHZ for trios¶
The De novo/CHZ for trios query identifies de novo and compound heterozygous variants in family trios and annotates each variant with the functional consequence and clinical impact.
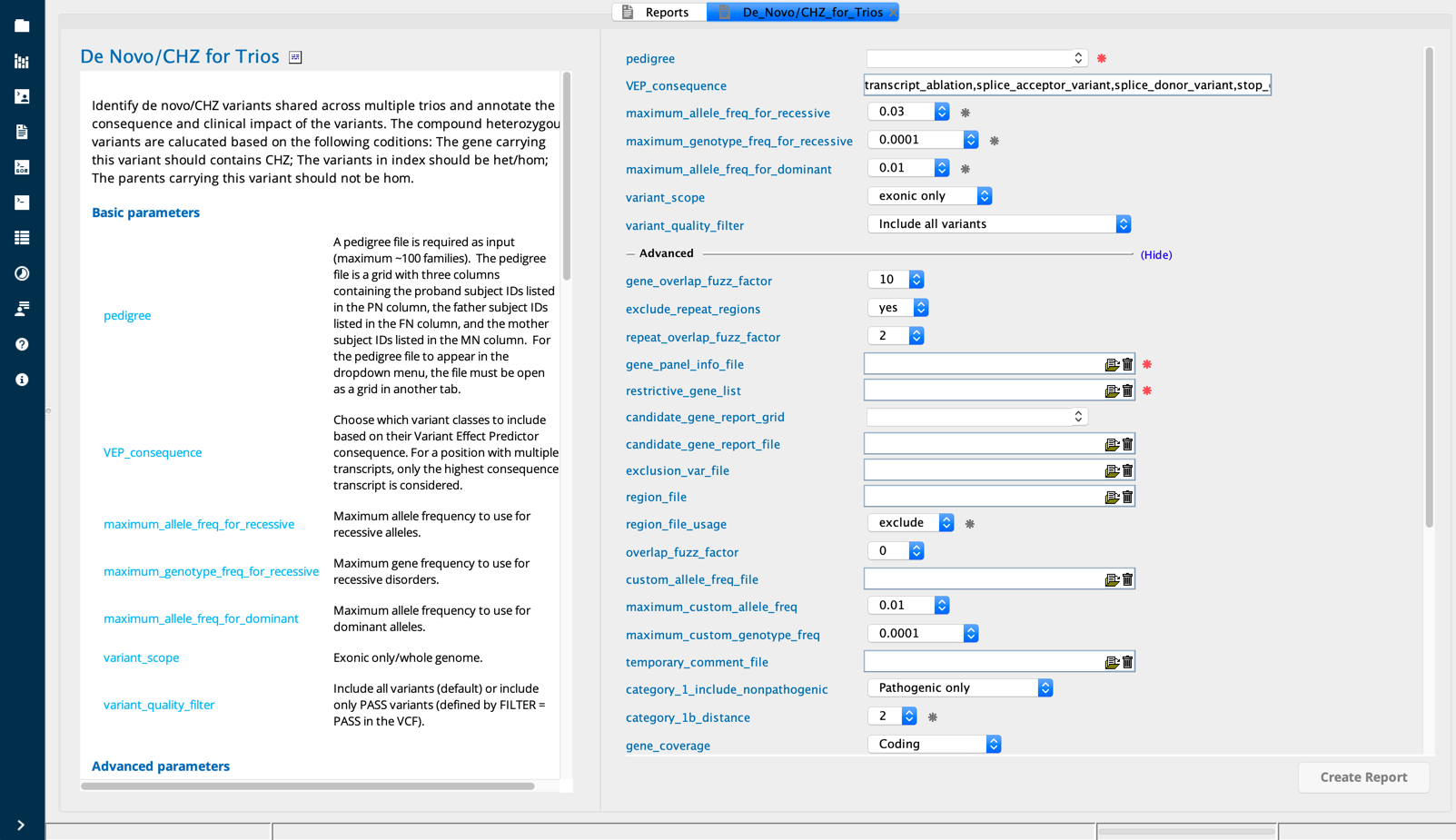
De Novo/CHZ for Trios in Sequence Miner¶
Example use case¶
The user has a pedigree file comprised of multiple trios, each comprised of one affected index case and unaffected parents. The user wishes to identify and annotate shared de novo variants and the genes carrying de novo variants across the index cases in all the families.
Description of the algorithm¶
This query annotates each de novo variant and compound heterozygous (CHZ) variants across index subjects. The user provides a pedigree file with three columns: PN (proband), FN (father), and MN (mother), listing one family per row. The steps to generate a pedigree file can be found in the De novo/CHZ for trios user manual.
Identification of variants in the index
First, variants that meet the user’s criteria for the following filters are extracted for each index:
minGLlikelihoodscore > 5 (or > user defined minimum)MinReadDepth> 8 (or > user defined minimum)VarFilterfor quality (PASS or include LOW quality)
AND
are homozygous with
CallCopies= 2 andCallRatio>= 0.66 (or CallRatio > user defined minimum ratio for homozygous call)
OR
are heterozygous with
CallCopies= 1 andminHetCallPerc>= 0.2 andCallRatio<= (1.0 - 0.2) (orCallRatiobetween the user defined minimum and (1.0 -minHetCallPerc)).
The filter for MinReadDepth and the others described above apply only to those in the index. This step defines the qualifying variants for further assessment in the parents.
Assessment of the above variants in the parents
deNovo assignment: Variants that meet the above filtering criteria are called “deNovo” if all of the following additional criteria are met:
The index carries the variant
Neither of the parents carry the variant
In order to rule out the presence of the variant in the parents, the coverage for the variant locus in both parents meets the user defined threshold for read depth (
minReadDepth), genotype likelihood (minGTlikelihood), and call ratio (minHetCallPercandminHomCallPerc).
CHZ assignment: An index is called “CHZ” for variants that meet the above filtering criteria if all of the following additional criteria are met:
The index carries at least two variants in a gene (het or hom)
Each variant is contributed by a different parent
In order to confirm that each variant is contributed by a different parent, the coverage for the variant locus in both parents meets the user defined threshold for read depth (
minReadDepth), genotype likelihood (minGTlikelihood), and call ratio (minHetCallPercandminHomCallPerc).
Coverage (read depth) at the variant loci in the probands and parents is extracted from the VarCount column (the number of reads per variant) in the candidate_variants.gord file.
Interpreting the output¶
Genetic disorders arising from de novo variants are identified by screening for overrepresentation of de novo variants in the same gene(s) across index cases in unrelated families. Begin by sorting the sum_deNovo column to find those variants present in probands as de novo variants. Confirm the variant presence in the proband and absence in the parents by following the De novo/CHZ for trios user manual.
Column descriptions¶
Group |
Column |
Description |
|---|---|---|
Gene |
DmaxAf |
Maximum allele frequency for dominant alleles set as input parameter |
MOI |
Mode of inheritance (default is ‘All’ for autosomal and sex chromosomes) |
|
RmaxAf |
Maximum allele frequency for recessive alleles set as input parameter |
|
RmaxGf |
Maximum genotype frequency for recessive disorders set as input parameter |
|
Symbol |
Based on HGNC when it exists, otherwise it is the Ensembl internal alias |
|
KNOWN |
distance |
The distance between a known pathogenic variant (Cat1, pathogenic annotation in HGMD, ClinVar, or OMIM) and the identified variant |
exactMatch |
A Boolean column (1/0) indicating if the variant (chromosome, position, reference, call) is a direct match to a known pathogenic variant (instead of near a known variant, or at the same position with a different call allele) |
|
Set_MaxClinImpact |
The maximum reported clinical impact of the variant (pathogenic/unknown etc) as annotated by HGMD, ClinVar, and OMIM |
|
var_diseases |
Diseases known to be associated with the variant as annotated by HGMD, ClinVar, and OMIM |
|
Max |
AF |
Maximum reported allele frequency across the population surveys from 1000GP3, EVS, EXAC, Kyoto, GONL (Variant View - Frequencies) |
consequence |
VEP predicted consequence for a variant producing the the greatest impact on the transcript |
|
Impact |
Classification of the level of severity of the transcript consequence type assigned by VEP |
|
Score |
Maximum score for the variant as observed in dbNSFP [Score=max ((1-Sift_score), Polyphen2_HDIV_score, Polyphen2_HVAR_score)] |
|
Sum |
deNovo |
The number of input probands (PNs) carrying the variant in a “deNovo” state |
DIAG_CHZ |
The number of input probands (PNs) carrying compound heterozygous variants |
|
Other columns |
allCount |
The number of input probands (PNs) carrying the variant |
Amino_acids |
Reference amino acid/substituted amino acid (in the case of a missense variant) |
|
Biotype |
Biological class of transcript or regulatory feature |
|
CDS_postion |
Relative position of base pair in coding sequence |
|
Gene |
Ensembl stable ID of affected gene |
|
Protein_position |
Relative position of amino acid in protein |
|
Refgene |
Accession number from NCBI using lookup into Ensembl 69 using feature |
|
Transcript_count |
The number of transcripts overlapping this variant |