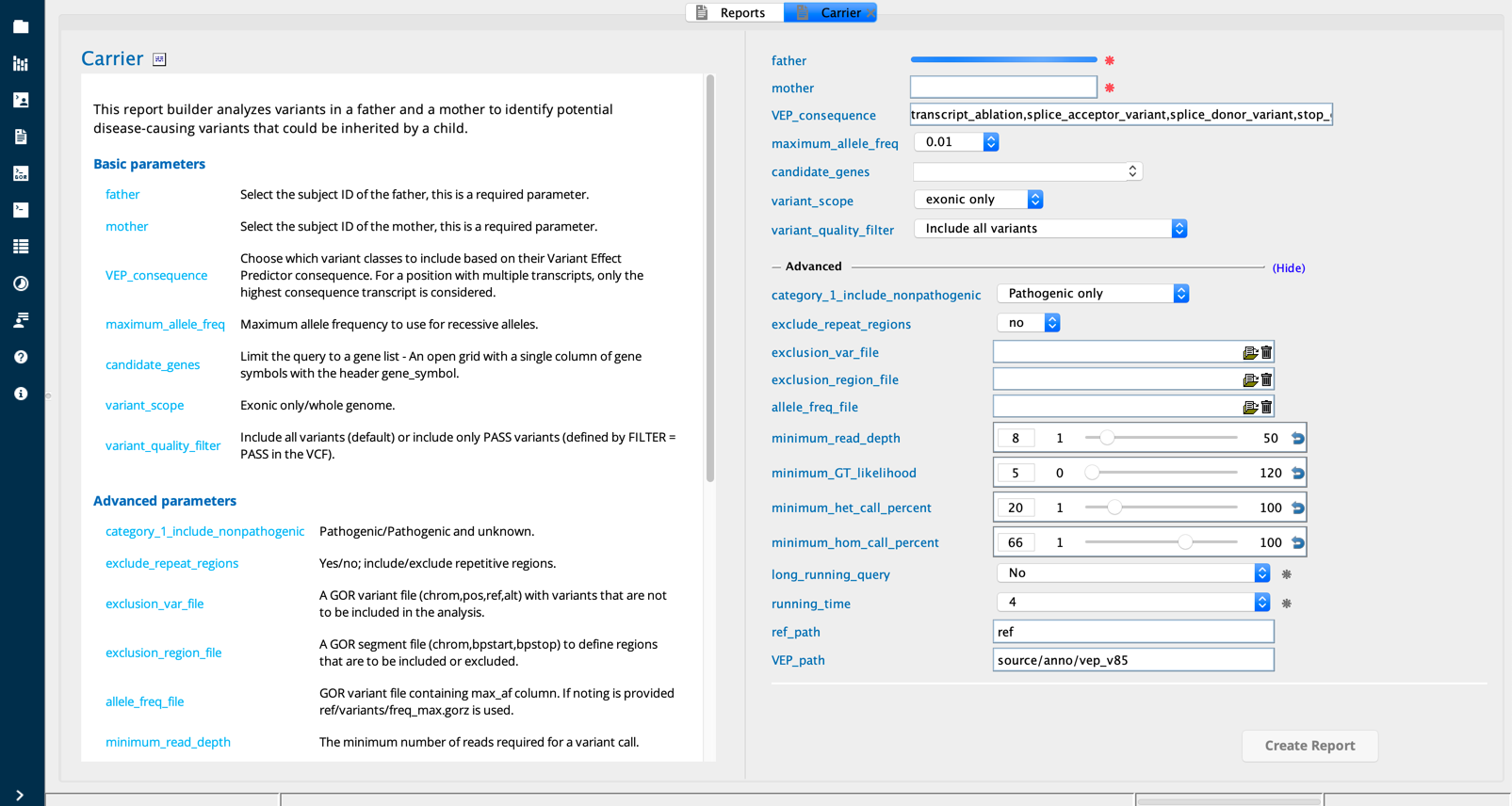
Carrier¶
The Carrier report builder analyzes variants in parents to identify potential disease-causing variants that are heterozygous in the parents and could be inherited by a child to produce a recessive disorder.
Carrier module in Sequence Miner¶
Example use case¶
The user wishes to screen whole exome sequencing (WES) reads from expectant parents to identify all sequence variants that may be inherited by their offspring. The user designates several filters to apply to the identified variants: a range of VEP consequences, maximum allele frequency, a target range of whole genome or exon-only, and sequence quality thresholds.
Description of the algorithm¶
The algorithm extracts all variants from the VEP whole exome file (source/anno/vep_v3-4-2/vep_single_wes.gord) that meet the user’s filtering criteria for max_consequence (maximum predicted consequence) and max_af (maximum allele frequency). Annotation from the clinical_variants.gorz file is joined to Known pathogenic variants in this subset in order to include the clinical database source (HGMD, ClinVar or OMIM in the Known_dbSource column), Gene_diseases, MaxClinImpact, and Var_diseases. Further annotation includes coverage-per-base and average coverage across a 10 base window (within +/- 5 bases adjacent to the variant).
Interpreting the output¶
The results are summarized in four different perspectives:
Default view - Results are presented in multiple columns of annotation including zygosity; VEP-predicted consequence, impact and score; genomic coordinates; descriptions; gene ontology ID; OMIM ID; etc.
InBothParents - This perspective presents only the subset of identified variants that meet the applied filters and that are present in both parents.
RecessiveCandidates - This perspective presents all variants that meet the applied filters and for which either or both parents are heterozygous.
RecessiveKnownVar - Recessive known variants are those present in either parent and that are reported in HGMD, ClinVar or OMIM as known Category 1 (pathogenic) variants.
Column descriptions¶
Group |
Column |
Description |
|---|---|---|
Basic |
CHROM |
|
POS |
||
Reference |
||
father |
CallRatio |
Proportion of reads containing the variation call; expected to be close to 0.5 for heterozygous calls and close to 1 for homozygous calls |
Depth |
The number of reads used in evaluating the corresponding call |
|
extCovAvgDepth10 |
The coverage depth extracted for variants and averaged across a 10 base window centered on the variant, meaning within 5bp on either side of the variant |
|
extCovDepth |
The depth of coverage at the variant nucleotide(s) |
|
FILTER |
Quality parameter using the ratio between gt-quality and depth showing if the call is considered of LowQual quality or acceptable quality (PASS); this is a crude quality measure |
|
formatZip |
VCF genotype field |
|
FS |
Fisher’s exact test of read strand; if the reference reads are balanced between forward and reverse strands, then the alternate reads should be as well |
|
genesum_het |
The sum of the number of heterozygous variants for a given gene |
|
genesum_hom |
The sum of the number of homozygous variants for a given gene |
|
genesum_miss |
The number of variants in the gene that have coverage less than 8 and are therefore excluded from the analysis |
|
GL_call |
A statistical measure indicating the likelihood that the call is wrong; the scale has been converted to use only integer numbers - the higher the number, the less likely that the call is wrong |
|
het |
Boolean for hereozygosity (1) or absence of heterozygosity (0) of the variant defined by a CallCopies value = 1 |
|
hetORhom |
“hom” for homozygosity and “het” for heterozygosity |
|
hom |
Boolean for homozygosity (1) or absence of homozygosity (0) of the variant defined by a CallCopies value = 2 |
|
miss |
A Boolean column set to 1 if the variant has coverage less than 8 |
|
PN |
Subject ID |
|
vcf_alt |
Alternate (variant) nucleotide sequence as reported in the vcf |
|
vcf_pos |
Coordinate (position) of the reference nucleotide sequence |
|
vcf_ref |
Reference nucleotide sequence |
|
Gene |
Aliases |
Aliases of the given gene |
biotype |
Biological class of gene |
|
cdsEnd |
cDNA end position |
|
cdsStart |
cDNA start position |
|
Description |
Description of the gene, i.e. full gene name |
|
gene_stable_id |
Ensembl stable ID for the gene |
|
Paralogs |
Paralogs of the given gene |
|
Pathways |
Biological pathway in which a given gene is found |
|
Strand |
Strand of the gene |
|
Symbol |
Based on HGNC when it exists, otherwise it is the Ensembl internal alias |
|
GO |
Description |
Gene ontology category description |
IDs |
Gene ontology identifiers |
|
KNOWN |
dbsource |
Variants known in one of the following databases: HGMD, OMIM and ClinVar |
Gene_diseases |
Diseases known to be associated with the gene as annotated by the dbsource column |
|
MaxClinImpact |
The maximum clinical consequence of the variant (pathogenic, unknown etc) annotated by any one of HGMD, OMIM or ClinVar |
|
var_diseases |
Diseases known to be associated with the variant as annotated by the dbsource column |
|
lis |
dbSourceMaxClinImpact |
|
disease |
||
mother |
Same columns as in father group |
Same as described in father group |
OMIM |
Descriptions |
OMIM disease descriptions for the gene |
IDs |
OMIM ID of the gene |
|
VEP |
Amino_Acids |
Amino acid with and without mutation (only provided if the variation affects the protein-coding sequence), otherwise “.” |
max_consequence |
VEP predicted consequence for a variant producing the the greatest impact on the transcript |
|
Max_Impact |
Classification of the level of severity of the transcript consequence type assigned by VEP |
|
Max_Score |
Maximum score for the variant as observed in dbNSFP [Score=max ((1-Sift_score), Polyphen2_HDIV_score, Polyphen2_HVAR_score)] |
|
Protein_Position |
Position of the amino acid in the protein sequence (only if the variant falls within a coding sequence); a value is given for each corresponding transcipt specified in the CDS position field |
|
Other columns |
Allele |
Alternative allele in VCF format |
DIAG_AMCGcat |
Categorization of the likelihood of pathogenicity of sequence variants according to the ACMG scheme; values range from cat1 (most severe) to cat4 (least severe); see AMCG categories below |
|
MAX_AF |
Maximum reported allele frequency across the population surveys from 1000GP3, EVS, EXAC, Kyoto, GONL` |
|
MaxClinImpact |
Max clinical impact from the clinImpact column |
|
Recessive |
Indicates whether the child has a high probability of being recessive for a variant or not (“true” or “false”); “true” means the father and mother are heterozygous for a variant or compound heterozygous for the gene |
|
set_pathway |
List of Ensembl pathways annotated for this gene from BIOCYC,HGMD,KEGG, Pathway Interaction Database (PID), REACTOME, and WikiPathways |
AMCG categories¶
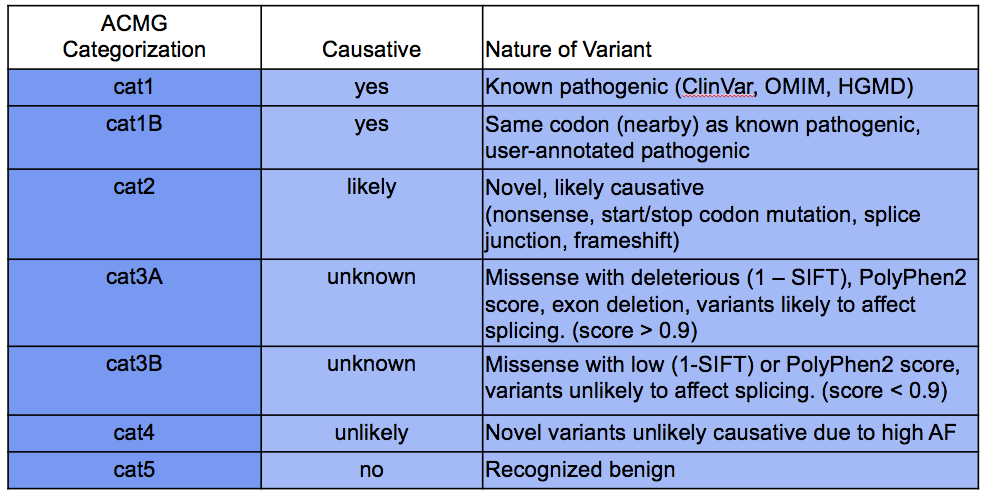
AMCG categories¶
Perspective views¶
The Default view perspective lists all variants. Additional perspectives focus on subsets of the columns in the default view.
Perspective |
Description |
|---|---|
Default view |
Lists all variants |
InBothParents |
The variant is categorized as “recessive” (condition 1); the variant is heterozygous or homozygous in both father and mother (condition 2); the gene is related to known disease (condition 3); (Recessive = ‘true’ and FATHER_hetORhom != ‘’ and MOTHER_hetORhom != ‘’ and (len(KNOWN_Gene_diseases)>1 or len(OMIM_Descriptions)>1)) |
RecessiveCandidates |
The variant is categorized as “recessive” (condition 1); the gene is related to a known disease (condition 2). This criterion is less stringent than InBothParents (Recessive = ‘true’ and (len(KNOWN_Gene_diseases)>1 or len(OMIM_Descriptions)>1)) |
RecessiveKnownVar |
The variant is categorized as “recessive” (condition 1); the variant is a known pathogenic variant labeled ‘cat1’ (condition 2) |
Drill-in reports¶
Drill-in |
Description |
|---|---|
GeneVariantsInParents |
For the selected gene variant, this drill-in report lists all variants in the same gene that are carried by the parents |